投资要点
| 投资要点坚持源头创新,立足全球小分子赛道竞争:迪哲医药致力于开发具备差异化竞争优势的全球创新药物,坚持源头创新,专注于新靶点的发现和作用机理验证。目前,公司在研管线6个产品,其中舒沃替尼是全球唯一全线获FDA与CDE突破性疗法认定治疗EGFR Exon20ins 突变型晚期非小细胞肺癌(NSCLC)且同时在中国获批NSCLC相关适应症的酪氨酸激酶抑制剂(TKI),戈利昔替尼是全球首个且唯一获FDA快速通道认定针对外周T细胞淋巴瘤(PTCL)且在中国获批上市用于治疗复发/难治PTCL的JAK1抑制剂。国内商业化进展顺利,美国顺利报产:根据弗若斯特沙利文测算,预计到2024和2030年,全球的EGFR Exon20 ins的非小细胞肺癌新发患者人数将分别达到7.4万和8.6万人,中国新发患者人数将达到3.5万和4.2万。小分子靶向药治疗空白,舒沃替尼2023年8月国内获批上市,2024年销售额接近4亿元,我们预计国内2025-2026年销售额为7亿和12亿元;美国FDA也受理了NDA,并授予了优先审评资格,我们预计2025H2有望美国获批上市,有望填补海外口服小分子药物治疗空白。差异化适应症设计,美国注册加速中:戈利昔替尼是全球首个且唯一针对外周T细胞淋巴瘤(PTCL)的高选择性口服JAK1 抑制剂,临床表现卓越,安全性和耐受性良好,有望冲破PTCL治疗瓶颈。2024年6月实现了国内获批上市,美国有望今年报产,是潜在同类最佳的针对PTCL的口服小分子靶向药。全球首创,攻克BTKi和EGFR TKI耐药困境:DZD8586和DZD6008是全球首创的靶点/结构。临床前和临床初期结果展示了惊艳的抗肿瘤效果,安全性良好,今年有望国内进入到注册临床阶段。有望填补当前未被满足的临床药物空白。盈利预测与投资评级:四个产品国内销售峰值我们预计达到近60亿人民币(2032年)。舒沃替尼和戈利昔替尼海外销售峰值我们预计分别达到9亿美金(2031年)和6亿美金(2031年),暂不考虑DZD8586和DZD6008的海外销售。采用FCFF估值法:公司的四款产品的绝对估值总数为343亿人民币,对应目标股价为82.26元/股。采用分步计算的P/S和P/E相对估值法,公司目标市值362亿元。公司国内商业化推进顺利,产品出海确定性高,在研品种潜力大,首次覆盖给予“买入”评级。风险提示:新药研发及审批进展不及预期;药品的销售不及预期;政策影响对产品价格的不确定性;海外授权不及预期。 |
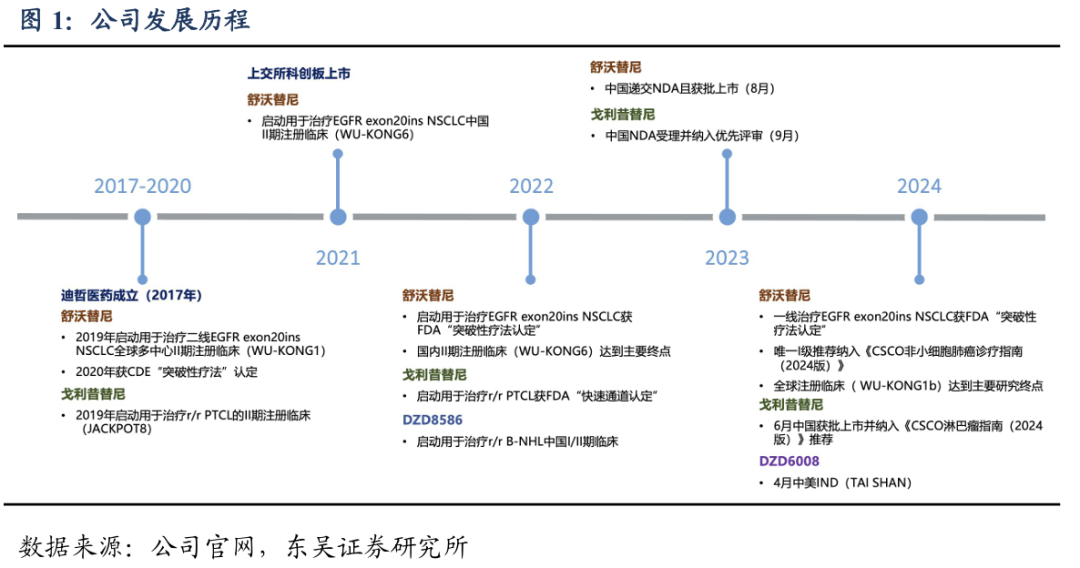
1.坚定自主研发路线,迈向全球的中国创新药企
迪哲医药成立于2017年,2021年12月成功在科创板上市,是一家具备全球竞争力的创新驱动型生物医药公司。公司致力于开发具备差异化竞争优势的全球创新药物,坚持源头创新,专注于新靶点的发现和作用机理验证。目前,公司已经建立起具备全球竞争力的研发管线,其中舒沃替尼是全球唯一全线获FDA与CDE突破性疗法认定治疗EGFR Exon20ins 突变型晚期非小细胞肺癌(NSCLC)且同时在中国获批NSCLC相关适应症的酪氨酸激酶抑制剂(TKI),戈利昔替尼是全球首个且唯一获FDA快速通道认定针对外周T细胞淋巴瘤(PTCL)且在中国获批上市用于治疗复发/难治PTCL的JAK1抑制剂。
1.1.顶级跨国医药公司高管团队,深厚积淀推动公司突破前行
公司的核心管理团队具有十余年的医药行业工作经验,拥有丰富的全球创新药领域管理、研发和商业化经验。公司董事长兼总经理张小林博士是北京大学分子医学研究所客座教授和哈佛大学医学院癌症中心的博士后,曾在阿斯利康担任全球副总裁并建立中国创新中心。首席医学官杨振帆博士拥有超过20年的临床实践和药物研发经验,曾在阿斯利康中国创新中心担任项目总监和医学总监。首席商务官吴清漪女士拥有20多年商业化销售经验,曾在拜耳、礼来亚洲、辉瑞和阿斯利康等跨国药企任职,曾任百济神州大中华区首席商务官。这些资深团队成员的多元背景和专业知识为公司的高质量发展提供了坚实的支持。

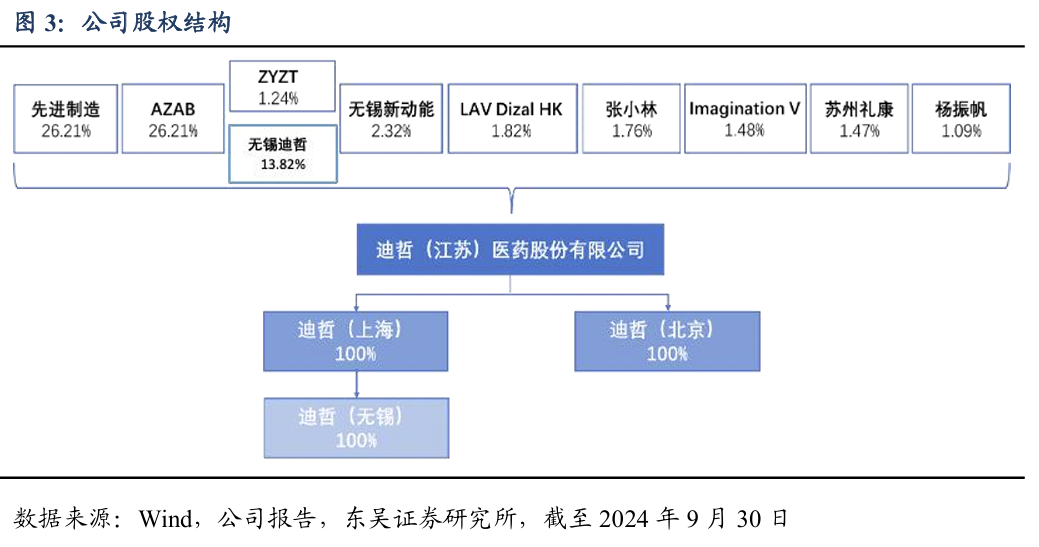
公司的股份主要集中于先进制造和AstraZeneca AB两大股东,无控股股东及实际控制人。截至2024年9月30日,两大主要股东持股比例均为26.21%。先进制造是由国投创新投资管理有限公司旗下基金管理,AZAB是阿斯利康的全资子公司。无锡迪哲与ZYTZ为迪哲的员工持股平台,合计持股15.06%,董事长张小林为ZYTZ与无锡迪哲执行事务合伙人无锡敦禾的最终实际控制人。公司共有三家全资子公司(包括间接子公司),其中迪哲上海主要从事医药行业内的技术开发等业务,迪哲北京则负责药品批发等工作,而迪哲无锡作为迪哲医药的间接子公司,则负责药品生产等工作。
1.2.坐拥核心技术平台,在研管线进入收获期
研发架构持续完善,六大技术平台形成完整创新药物研发体系。公司具备行业内领先的转化科学研究能力和技术平台,为新药研发立项提供了重要支持。此外,公司也自主建立了完整的创新药研发平台,覆盖了从药物靶点发现和机理验证,到转化医学研究,再到化合物分子设计与筛选,临床前研究,CMC,临床方案设计与执行等各个环节,在技术先进性和技术平台完整性方面具有较强竞争力。
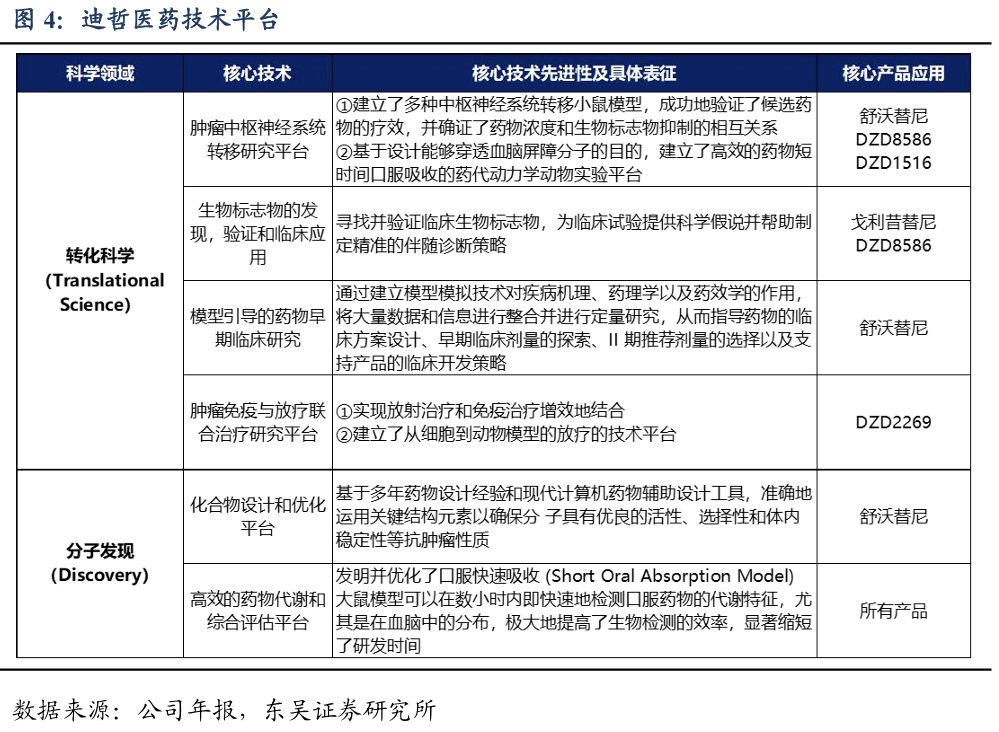
在研管线集中于恶性肿瘤治疗领域,集中在肺癌和血液瘤。所有产品均享有完整的全球权益,并采用全球同步开发的模式。公司拥有6款具备全球竞争力的产品管线,4款产品已实现临床端的概念验证,2款产品处在全球关键性临床试验阶段。其中,公司的核心产品舒沃替尼、戈利昔替尼已在中国获批上市,DZD8586、DZD6008、DZD2269、DZD1516处于国际多中心临床研究阶段。公司还储备了多个处于临床前研究阶段的候选创新药物,后续将继续聚焦在小分子治疗领域,以推出全球首创(First-in-class)和具有突破性潜力的治疗方法为目标。
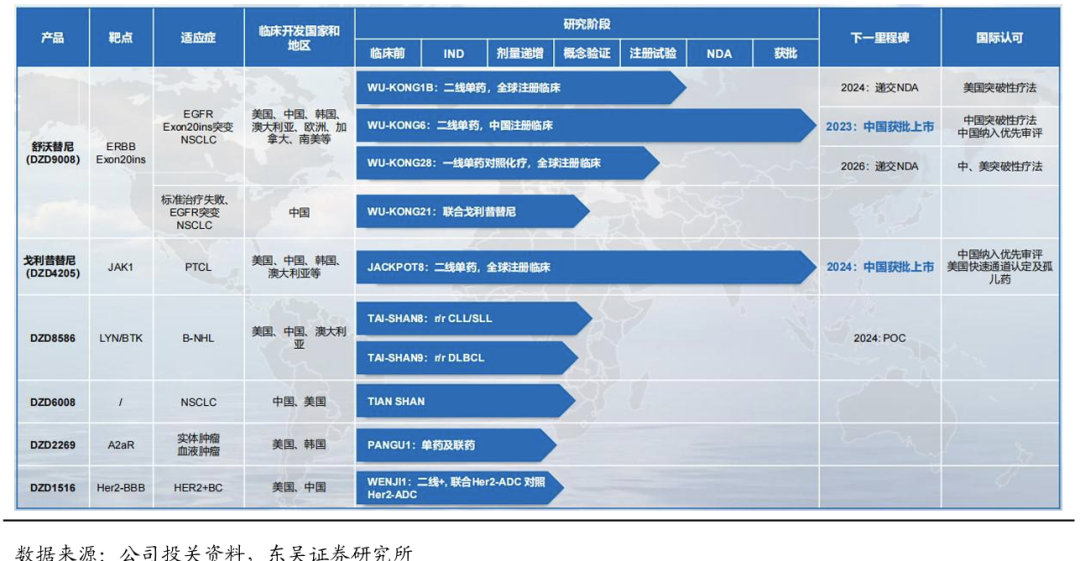
1.3.研发投入持续增加,商业化布局静候业绩兑现
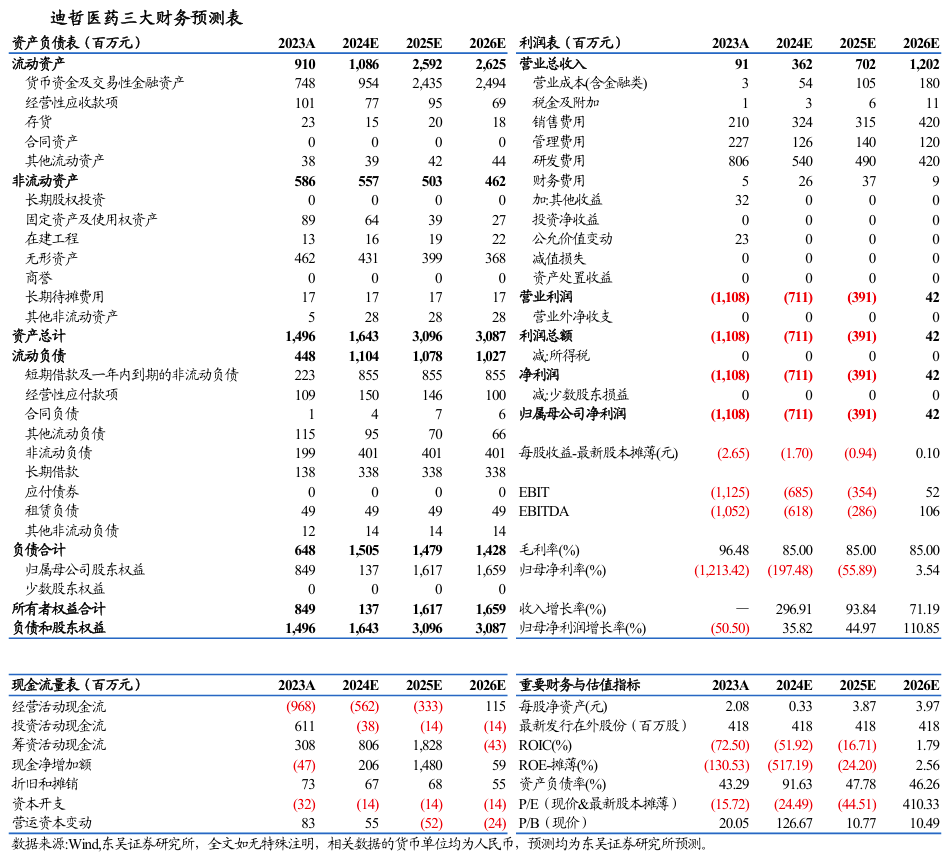
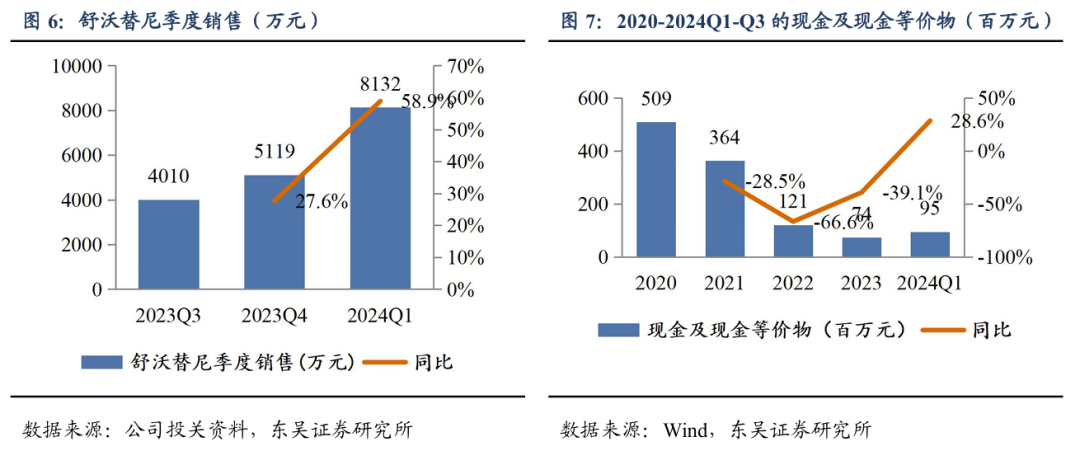
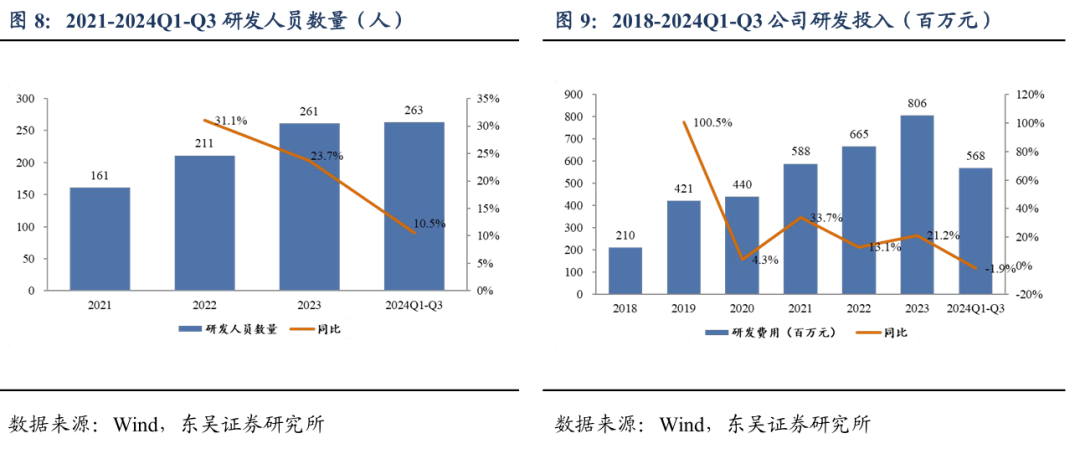
舒沃替尼(舒沃哲)于2023年8月批准上市,2023年度销售收入9128.86万元,2024年Q1/Q2/Q3销售收入分别为8131.86万元/1.22亿元/1.26亿元,环比+59%/+50%/+3%。截至2024年9月30日,公司拥有现金及现金等价物7400万元。公司的研发投入由2018年2.10亿元提升至2023年8.06亿元,2024年Q1-Q3研发投入为5.68亿元,加速推进在研产品临床开发进度。截止到2024Q3,公司在全球10+国家拥有150+临床研究中心,公司研发人员263人,占比32.59%。
公司已建立一支专注于肺癌和血液瘤的商业化团队,构建了遍及全国的销售网络。2024年11月28日,舒沃替尼和戈利昔替尼成功纳入国家医保药品目录,将于2025年1月1日起正式实施。公司定位于参与全球化竞争,将与美国、欧盟等海外药品监管机构积极沟通,加快海外上市进程。
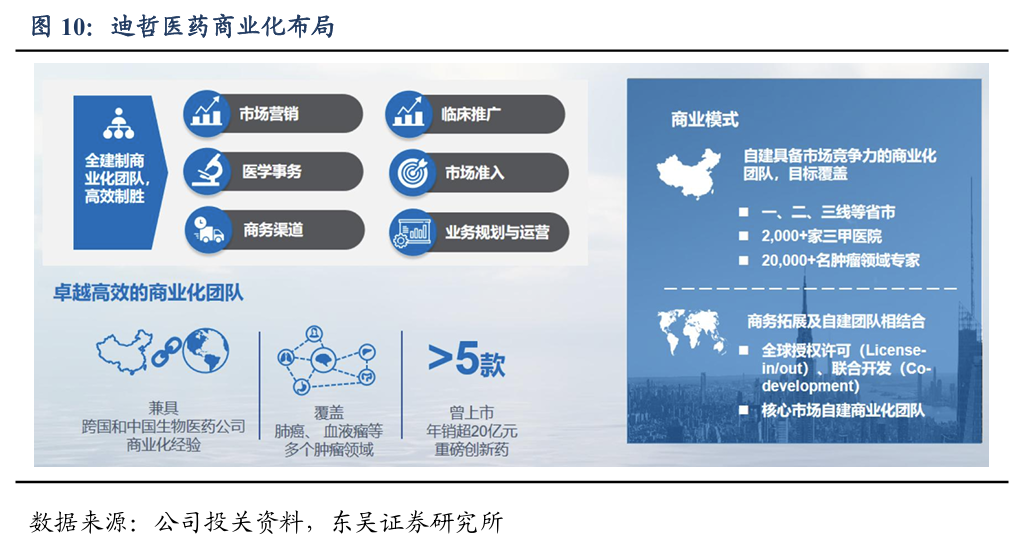
2.舒沃替尼:瞄准全球最佳EGFR-TKI药物
舒沃替尼是公司自主研发的特异性表皮生长因子受体酪氨酸激酶抑制剂(EGFR-TKI),针对 EGFR 20 号外显子插入突变(Exon20 ins)设计,是迄今为止肺癌领域全球首个且唯一全线治疗该适应症中、美双“突破性疗法认定”的大满贯创新药,同时对包括 Exon20ins 突变在内的多种EGFR突变都有较强活性,并保持对野生型EGFR高选择性。
2.1.针对Exon20 ins传统疗法效果不尽人意,新药破局需求大
据世界卫生组织统计,全球每年新增的肺癌患者约220万例,非小细胞肺癌(NSCLC)约占患者数的80-85%,这其中有32%的NSCLC患者被诊断为携带EGFR突变(在亚裔人群中及高加索人群中分别为30%-50%和10%-20%)。被诊断为EGFR突变的NSCLC患者中,约10%(4-12%)的患者携带20号外显子插入(Exon20 ins)突变。
EGFR exon20 ins主要是在 EGFR基因第20号外显子框架中进行复制或插入,它们具有高度的异质性,且大部分出现在C-螺旋环后面的环状结构中。同时由于EGFR exon20 ins与EGFR野生型在结构上的高度相似性,导致EGFR exon20 ins NSCLC患者对普通EGFR-TKIs具有天然的抗药性。除此之外,EGFR exon20 ins NSCLC患者肿瘤细胞的突变负载很小,即对免疫疗法的敏感度也较低。因此,亟需研制出更高效、更安全的新药。

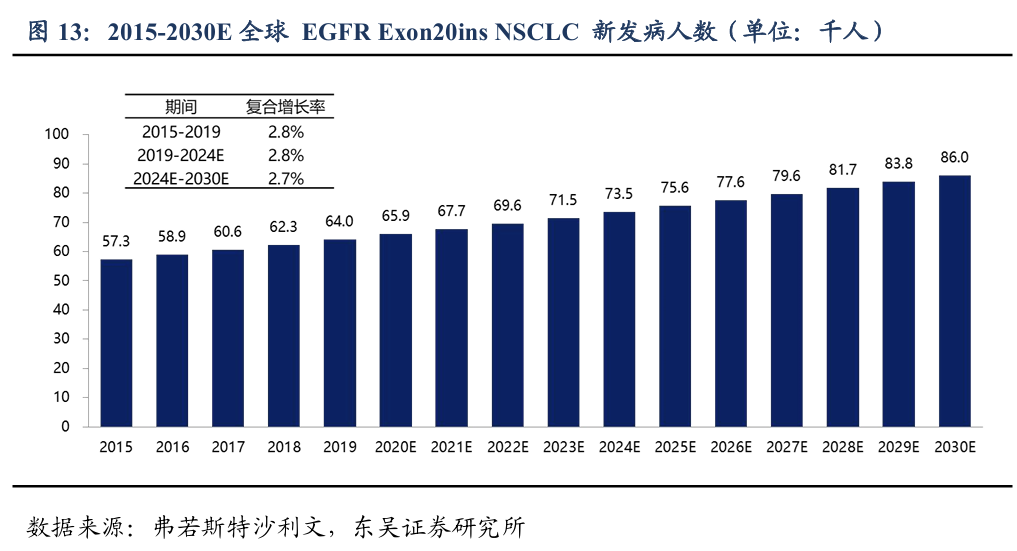
根据弗若斯特沙利文测算,从2015年到2019年,全球EGFR Exon20ins的非小细胞肺癌新发患者的数量从5.7万增加到6.4万,年复合增长率为2.8%。我们预计到2024和2030年,全球的EGFR Exon20 ins的非小细胞肺癌新发患者人数将分别达到7.4万和8.6万人。
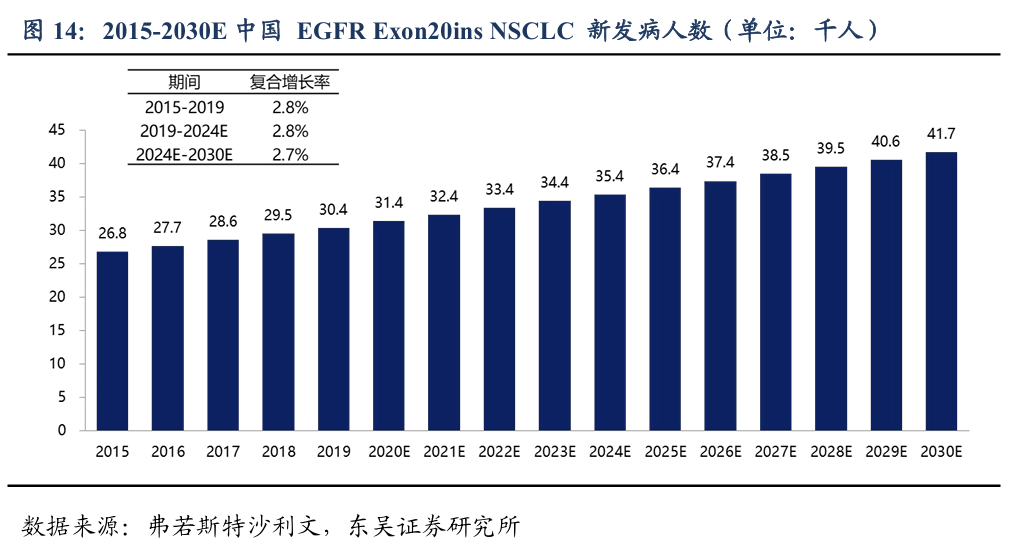
中国肺癌新发患者数量呈稳步增长趋势。从2015年到2019年间,中国EGFR Exon20 ins的非小细胞肺癌新发患者的数量从2.7万增加到3.0万,复合年增长率为2.8%。由于衰老、吸烟等风险因素的持续恶化,非小细胞肺癌新发患者的人群将继续扩大。弗若斯特沙利文预测,2024年中国的EGFR Exon20 ins非小细胞肺癌新发患者人数将达到3.5万。到2030年,这一数字预计增至4.2万。
2.2.舒沃替尼管线快速推进,开发历程屡创辉煌
源头结构设计差异化,高选择性和透脑性:舒沃替尼的设计思路以奥希替尼骨架结构为起点,将奥希替尼上旋转弹性较差的甲基吲哚替换为嘧啶铰链结合基序C-4上更灵活的苯胺苯基部分,并对苯环的邻位取代基团进行了进一步优化,在达到了高度靶向EGFR Exon20 ins的同时,保持对野生型EGFR的高选择性和对包括T790M在内的敏感突变(19和21号外显子突变)的有效活性。
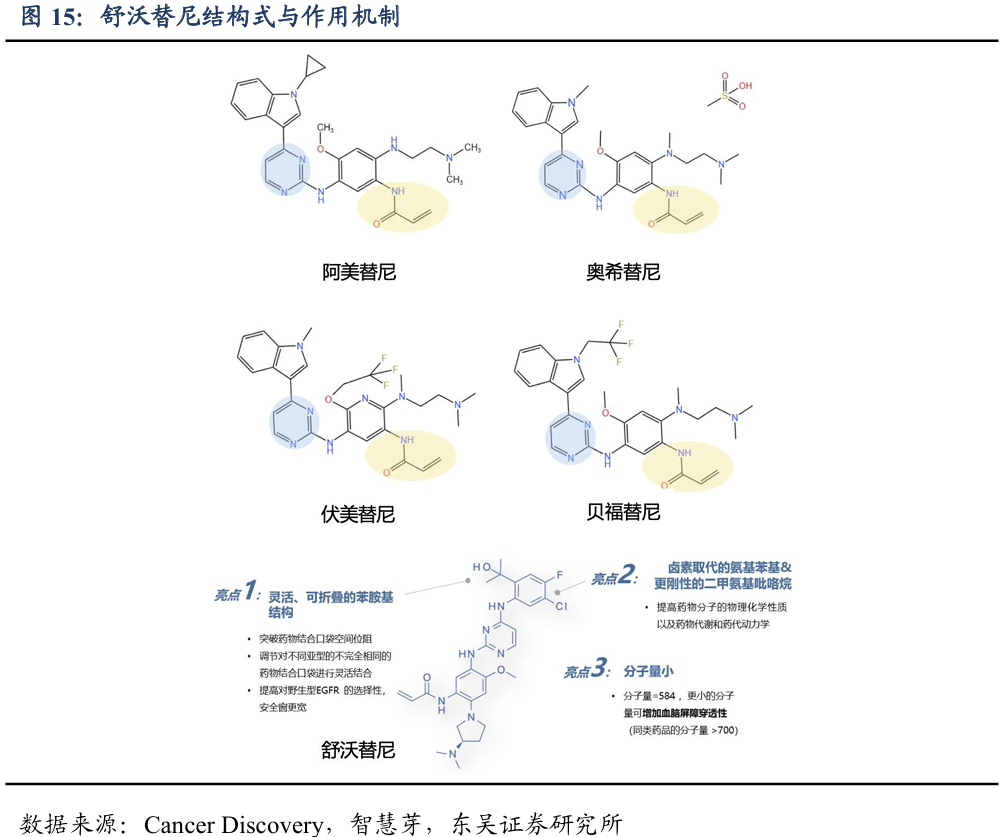
对野生型EGFR选择性更高,对Exon20 ins突变效果更好。临床前数据显示:舒沃替尼能够较强地抑制EGFR Exon20 ins,而对野生型EGFR抑制作用较弱。通常标准化IC50值越低,意味着TKI对该类突变抑制效果越强,而舒沃替尼对野生型EGFR有3~50倍的选择性,这一特性使得舒沃替尼在临床上有望获得更高的安全窗,降低不良事件的发生率和严重程度。同时舒沃替尼人体半衰期较长,达到约50小时左右,且PK曲线更平缓,更有利于每日一次口服给药及降低由于药物峰值浓度过高带来的不良事件发生。1-3代EGFR TKI在EGFR Exon20 ins患者中疗效不佳,有研究表明Exon20ins相较于敏感突变对于1-3代TKI 的敏感度平均低100倍左右。例如,中国回顾性临床研究表明在Exon20ins患者中应用奥希替尼,无论是原剂量或剂量翻倍,疗效欠佳(mPFS约为2个月)。

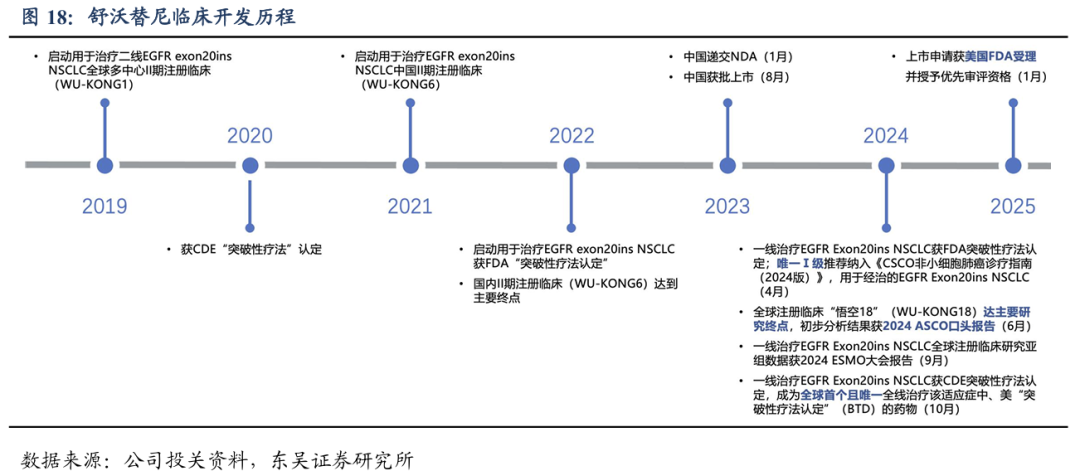
临床开发速度高效,美国商业化即将开启:舒沃替尼自进入临床开发阶段以来,连续三年在ASCO、WCLC、ESMO等国际顶级学术大会亮相并斩获三项口头报告。从国内首例临床研究受试者入组到正式获批上市,仅4年不到时间,突破了肺癌靶向药物临床开发新速度。海外临床试验推进速度也很快,25年1月美国NDA受理并授予了优先审评资格,我们预计2025H2有望实现美国获批上市。作为中国自主研发的首款针对EGFR Exon20 ins突变的肺癌靶向药,舒沃替尼是公司高效源头创新的最好证据,也体现了公司临床开发团队的高效和高标准。
2.3.舒沃替尼高效低毒,潜在同类最佳
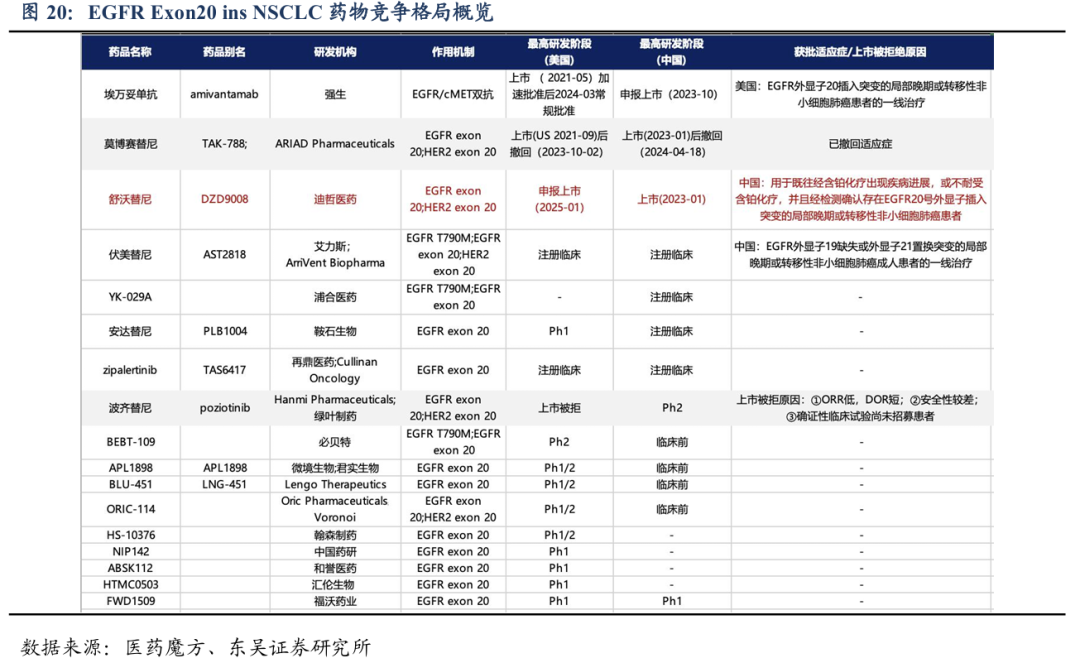
EGFR Exon20 ins是第三大EGFR突变,异质性强,常规EGFR-TKIs治疗无效,患者预后差。可采用聚合酶链式反应(PCR)和二代测序(NGS)进行检测。国内尚无一线治疗局部晚期或转移性EGFRex20ins NSCLC的靶向药物获批(武田的莫博赛替尼由于III期验证性临床失败已撤回该适应症),I级推荐方案建议参考无驱动基因晚期NSCLC的一线治疗,主要以PD-1单抗以及化疗为主。埃万妥单抗(Amivantamab)联合化疗已获美国食品药品监督管理局(FDA)批准用于一线治疗局部晚期或转移性EGFRex20ins NSCLC,国内尚未获批,暂获III级推荐。EGFRex20ins局部晚期或转移性NSCLC患者二线治疗,优先推荐靶向治疗药物舒沃替尼。

舒沃替尼先发优势明显,国内商业化进展顺利,美国有望在2025年获批上市。截至2025年1月,全球用于治疗EGFR Exon20 ins NSCLC的临床阶段在研药物有多款,已上市销售的药物仅有两款,分别为强生公司的埃万妥单抗和迪哲医药的舒沃替尼,分别于2021年5月和2023年1月在美国和中国获批上市。舒沃替尼国内商业化顺利,国内上市以来销售额逐季度环比增长,2024年销售额我们预计为4亿元左右。2024年底参加国家医保谈判成功纳入医保目录,2025年挂网价格为297元/150mg,我们预计12个月治疗费用约为21.68万元(降幅54%),体现了国家医保局对best-in-class药物的支持。舒沃替尼在美国也于2025年1月获得FDA的申报上市受理并获得优先审评资格,有望今年年底前获批上市,届时有望成为首个美国获批的国产肺癌靶向药。
Exon20 ins NSCLC的开发难度大:其中,莫博赛替尼由于临床III期研究失败主动撤市了,波齐替尼也由于有效性和安全性数据较差上市被CDE拒绝。目前,全球针对该适应症的竞争格局依然良好。后续在注册临床阶段的药物仅有来自艾力斯的伏美替尼、浦合医药、鞍石生物和再鼎医药的产品。舒沃替尼依靠先发优势,有望在此适应症上取得较大市占率。
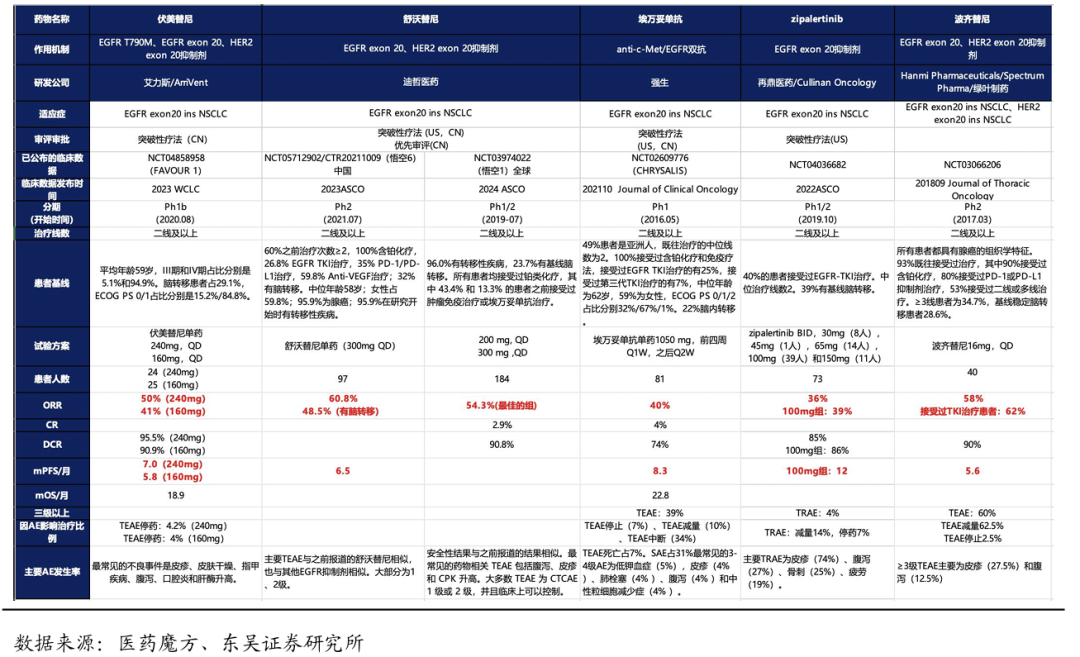
二线治疗中从有效性角度看,舒沃替尼的有效性数据接近埃万妥单抗。目前,针对Exon20 ins NSCLC的二线治疗的临床后期以及获批上市的创新药中,ORR在36%-61%之间,mPFS在5-8个月之间。非头对头临床数据显示:舒沃替尼(悟空6)相比已经上市的埃万妥单抗ORR更高(61% vs 40%),mPFS接近(6.5个月 vs 8.3个月),在既往接受过埃万妥单抗的患者中ORR高达67%,显示出舒沃替尼的独特优势。海外的临床中(悟空1)的患者基线中15%左右病人是前线埃万妥治疗失败的,舒沃单药的ORR仍能达到50%。
二线治疗中从安全性角度看,舒沃替尼的安全性明显优于埃万妥单抗。埃万妥单抗在目标剂量下,三级以上的不良反应高达39%,SAE发生率达到31%,停药的患者占7%,并且治疗期间有患者死亡。相比之下,舒沃替尼的安全性优势明显,与已获批上市的EGFR-TKI的安全性相似,绝大多数为1-2级不良反应。
从用药便捷度看,每日口服一次,患者依从性更高。埃万妥单抗需要每1-2周去医院静脉注射一次,比较麻烦。相比之下,舒沃替尼每日口服一次,更方便,患者依从性更好。

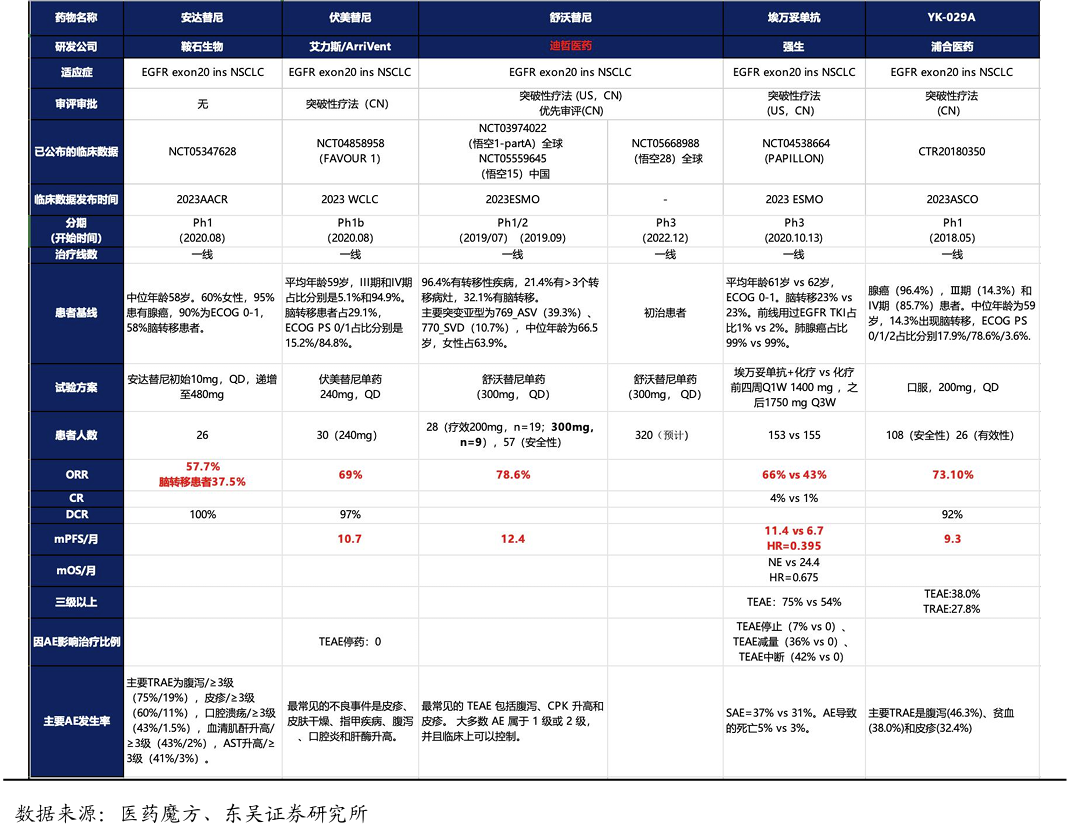
一线治疗中从有效性角度看,舒沃替尼的有效性数据优于埃万妥单抗。目前,针对Exon20 ins NSCLC的二线治疗的临床后期以及获批上市的创新药中,ORR在58%-79%之间,mPFS在9.3-12.4个月之间。非头对头临床数据显示:舒沃替尼单药相比已经上市的埃万妥单抗联合化疗的ORR更高(79% vs 66%),mPFS更高(12.4 vs 11.4个月)。舒沃单药相比联合疗法的优势明显,口服更加方便。

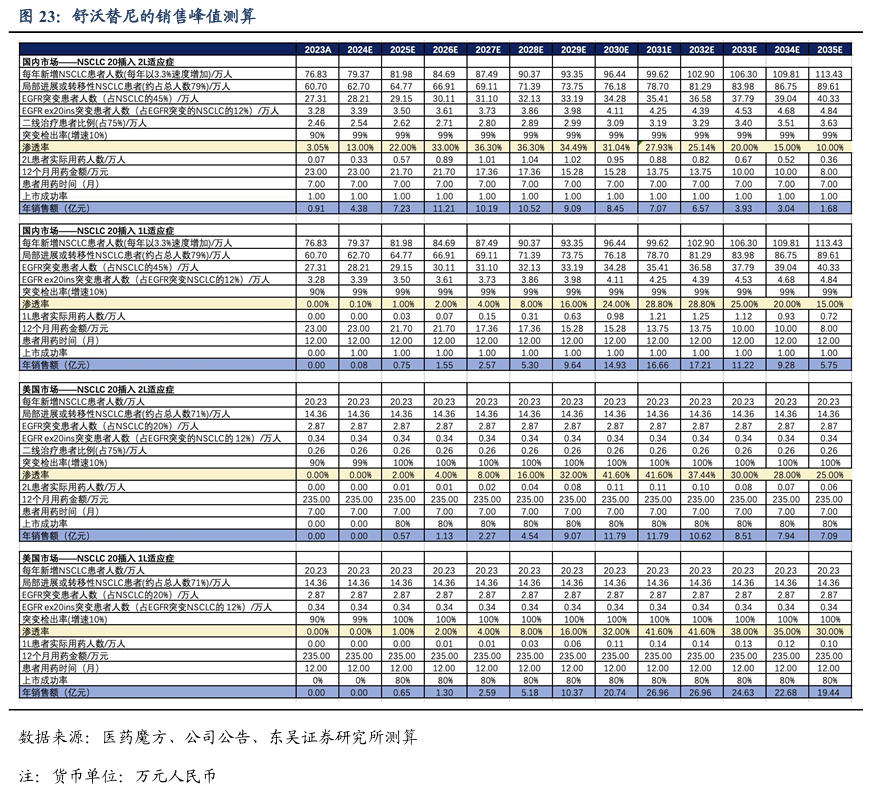
2.4.舒沃替尼峰值销售预测
上市时间假设:舒沃替尼国内已于2023年获批上市,我们预计美国2025年年底获批上市,欧盟、英国等海外国家我们预计2026年陆续获批上市。
药物降价幅度假设:舒沃替尼2025年首次执行医保价格,国内12个月的治疗费用约为21.7万元,之后我们预计每两年降价一次,终局价格维持在8万元/年。美国的治疗费用我们预计达到235万元/年。
渗透率假设:Exon20 ins NSCLC疗法有限,舒沃替尼的竞争格局良好,假设二线治疗峰值渗透率为36%;一线治疗的峰值渗透率有望达到29%。美国的渗透率有望达到40%左右。
基于上述假设,舒沃替尼国内销售峰值我们预计达到20亿元,海外销售峰值我们预计达到9亿美金(美国5亿美金)。
3.戈利昔替尼:全球首个且唯一治疗PTCL的JAK1抑制剂
戈利昔替尼(高瑞哲)是全球首个且唯一针对外周T细胞淋巴瘤(PTCL)的高选择性口服JAK1 抑制剂,从机制及临床结果验证了其具有PTCL全亚型获益的潜质。临床表现卓越,拥有较高且持续的客观缓解率,安全性和耐受性良好,有望冲破PTCL治疗瓶颈。此外,针对其它血液肿瘤、自身免疫性疾病等领域有望成为有力竞争者。戈利昔替尼正在中国、美国、韩国和澳大利亚等国家开展关键性临床试验。2022年2月获美国FDA的"快速通道认定"。2024年6月,国内获批上市用于治疗复发难治性外周T细胞淋巴瘤(r/r PTCL)。
3.1. PTCL患者人数逐年增加,生存期较低,缺乏有效治疗手段
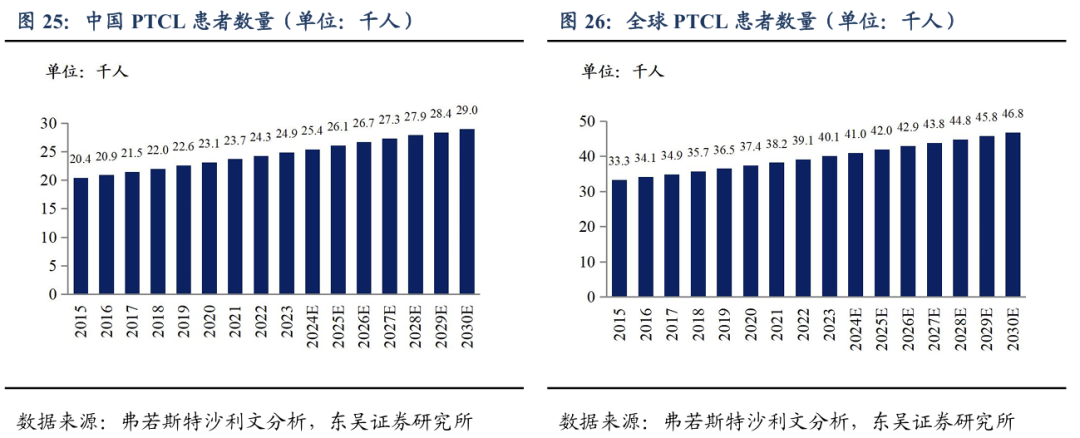
外周T细胞淋巴瘤(peripheral T cell lymphomas, PTCL)是一种异质性强、通常为侵袭性的非霍奇金氏淋巴瘤(NHL),占所有NHL的7%。整体预后差,一线治疗后复发率高。初治失败后复发难治性患者预后更差,其五年生存期低于30%。我国PTCL的发病率显著高于欧美国家,约占NHL的25%。
根据GLOBOCAN数据统计,2022年我国新增80,829例NHL患者,全球新增 553,010例NHL患者。据此测算,2023年我国约有24.9千人新增PTCL患者,全球约有40.1千人新增PTCL患者。根据弗若斯特沙利文的分析,PTCL患者2024年至2030 年的年复合增长率为2.2%,预计2030年我国和全球新发病例分别会达到29千人和46.8千人。
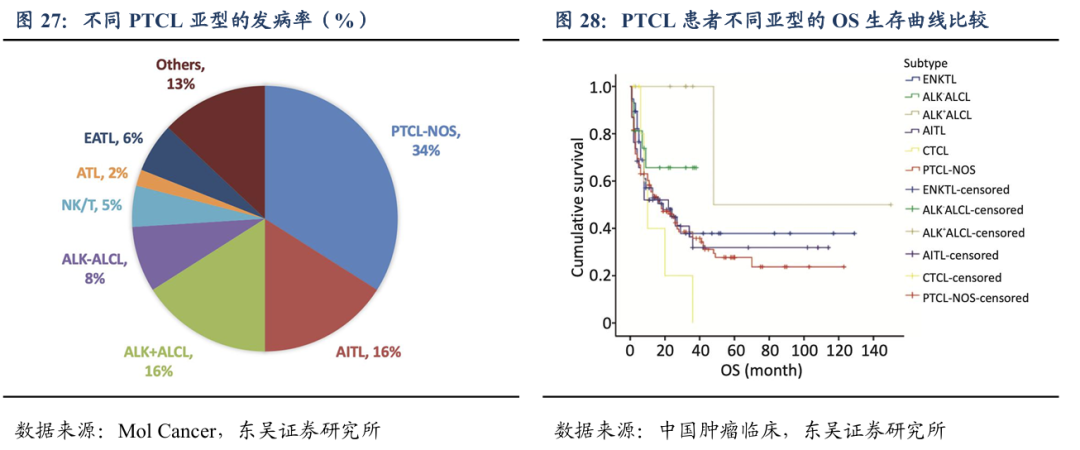
外周T细胞淋巴瘤(PTCL)侵袭性强、预后较差,依靠组织病理学和免疫组化明确诊断。PTCL包含至少29种亚型,最常见的是外周T细胞淋巴瘤-非特指型(PTCL-NOS)、血管免疫母T细胞淋巴瘤(AITL)、间变大细胞淋巴瘤(ALCL)、NK/T细胞淋巴瘤(NKTCL)和成人T细胞淋巴瘤/白血病(ATLL)。
3.2. PTCL近十年没有创新药上市,戈利昔替尼突破治疗瓶颈
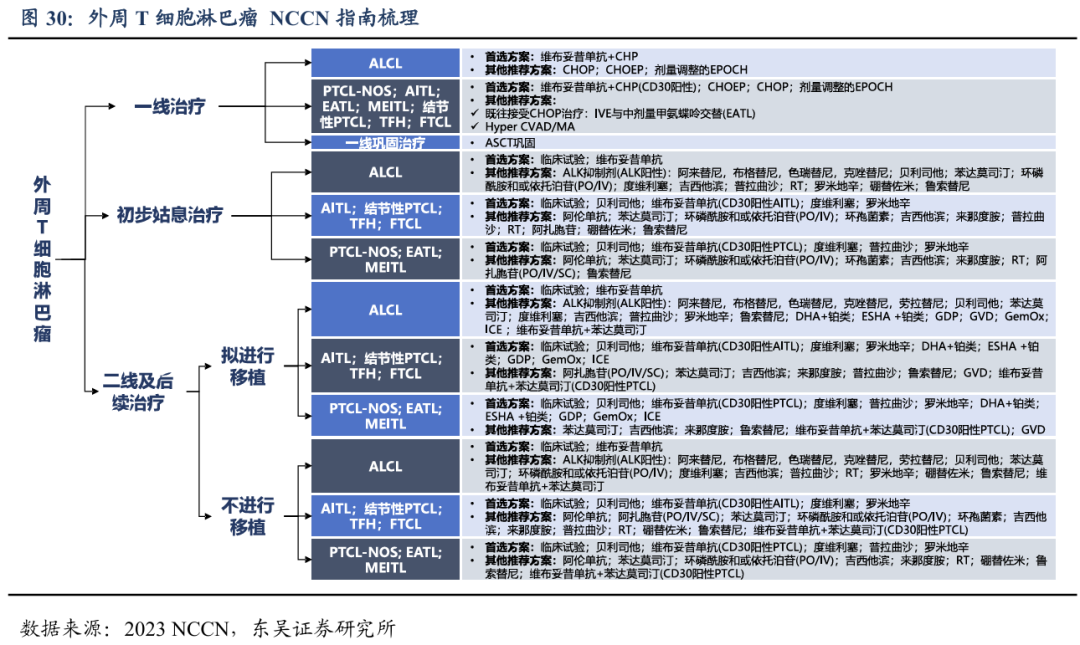
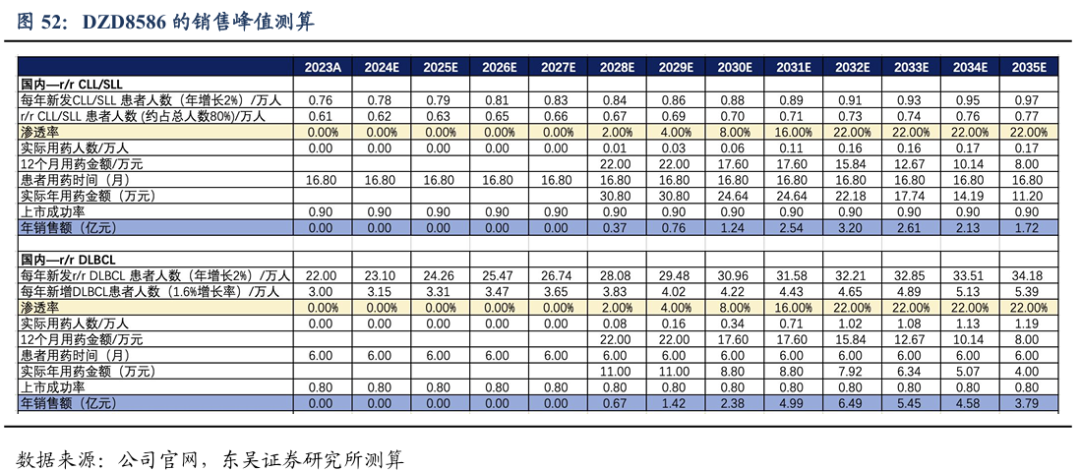
PTCL不同亚型的mOS仅为20个月左右,复发后治疗效果极为有限,3年生存率仅23%,OS仅5.8个月,该领域近十年没有创新药上市,目前尚缺乏非常有效的治疗方法。针对初治的PTCL患者以CHOP样的四药化疗方案为主。根据《CSCO淋巴瘤诊疗指南(2024版)》,PTCL一线治疗方案主要包括:维布妥昔单抗(CD30-MMAE ADC)联合CHP(环磷酰胺+阿霉素+强的松)、CHOP(环磷酰胺+阿霉素+长春新碱+泼尼松)、CHOEP(环磷酰胺、多柔比星、长春新碱、依托泊苷、强的松)和EPOCH(依托泊苷、强的松、长春新碱、环磷酰胺、多柔比星)。如果有ALK或者CD30等驱动基因阳性的患者优先选择ALK抑制剂或CD30 ADC。仅有少数患者经上述化疗后获得良好生存,大部分PTCL患者仍然预后不佳。对于复发或难治性(r/r)的PTCL,5年生存率不足30%,选择使用二线治疗方案,同样首选临床试验,其它单药方案的选择有西达本胺(HDAC抑制剂)、普拉曲沙(叶酸拮抗剂)、克唑替尼(ALK抑制剂)和苯达莫司汀(双功能基烷化剂)等。戈利昔替尼2024年首次获2024 CSCO指南的Ⅱ级推荐,用于治疗r/r PTCL患者。

美国国立综合癌症网络(NCCN)临床实践指南中针对PTCL的治疗方案和国内的CSCO指南相似,一线治疗药物也是维布妥昔单抗和多种化疗联合方案。二线及后续治疗首选药物主要包括:维布妥昔单抗(CD30 ADC)、ALK抑制剂、贝利司他(PI3K/HDAC抑制剂)、度维利塞(PI3K抑制剂)、罗米地辛(HDAC抑制剂)等。
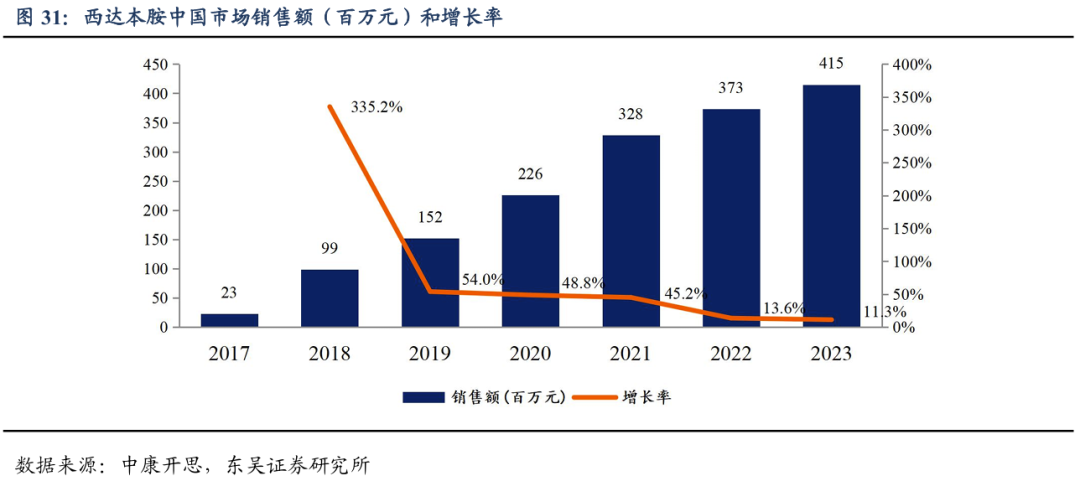
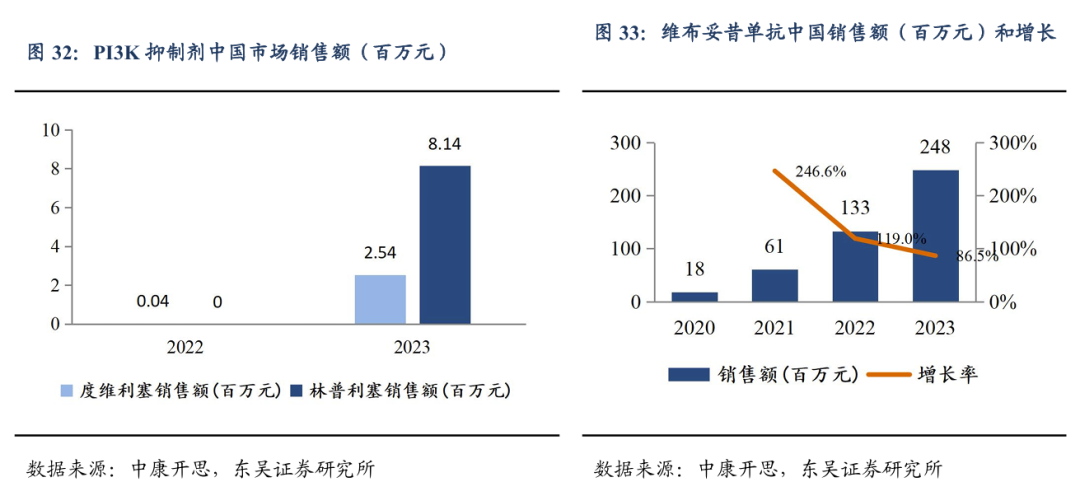
国内针对r/r PTCL 获批上市的创新药较少,在FDA近年来批准的 PTCL治疗药物中,使用较多的贝利司他还暂未在中国上市。目前国内获批的单药治疗药物为 HDAC 抑制剂西达本胺,叶酸拮抗剂普拉曲沙以及 PI3Kδ抑制剂度维利塞和林普利赛。目前,上市较早的西达本胺占领国内复发难治性 PTCL 药物主要市场份额。根据中康开思数据库测算,2023年西达本胺国内销售额为4.2亿元,同比增长11%。维布妥昔单抗2020年5月在国内上市,2023年国内销售额约为2.5亿元。PI3K抑制剂近两年陆续在国内上市,销售体量还较小。普拉曲沙2024Q1国内正式获批开始商业化。
3.3.戈利昔替尼针对r/r PTCL 潜在同类最佳
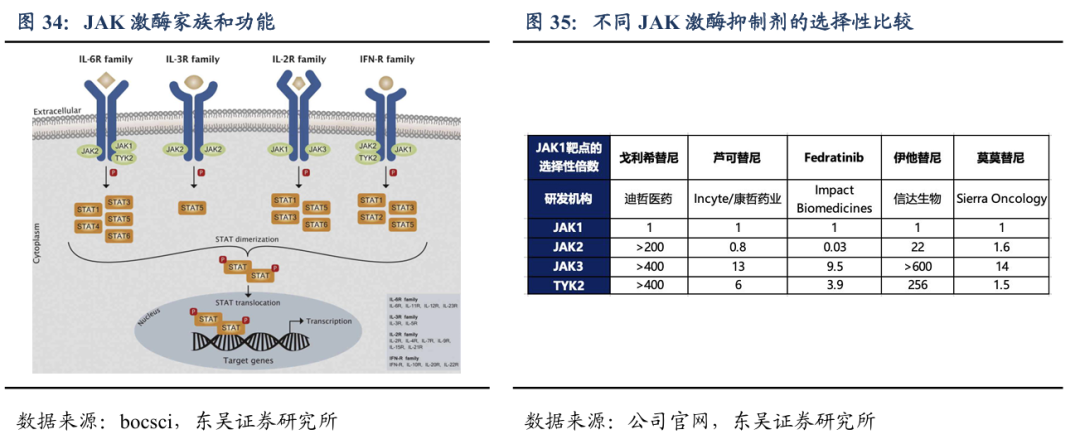
JAK是一种非受体型酪氨酸蛋白激酶,JAK激酶家族的蛋白有4个,分别为JAK1、JAK2、JAK3、TYK2,不同的受体结合选择不同的JAK蛋白。JAK1广泛存在于体内各种组织和细胞中,与IL-6、IFN等炎症因子的激活密切相关,是免疫、炎症等疾病领域的重要靶点。不同的JAK激酶亚型由不同的细胞因子激活,目前多数JAK抑制剂选择性较差,一般对多个JAK 激酶都具有抑制作用。由于现有的大多数非选择性JAK抑制剂均对JAK2有抑制作用,因此容易产生贫血等较大的毒副作用。戈利昔替尼的设计原则是提高对特定JAK1亚型的选择性抑制(大于200倍),避免由于抑制 JAK2带来的毒副作用,在保持药物对特定疾病疗效的同时降低不良反应。
戈利昔替尼是首个且唯一获FDA快速通道认定针对PTCL的国产创新药,2023 年 9 月,基于全球注册临床研究(JACKPOT8 B)结果递交中国新药上市申请(NDA)获受理并被纳入优先审评,2024年6月国内获批上市。临床研究结果连续4年获6项国际顶级学术大会口头报告,JACKPOT8 B 数据发表于 Lancet Oncology(IF=51)。
截至2025年1月,美国用于治疗PTCL的上市小分子靶向药和ADC仅有4款,分别是普拉曲沙(DHFR)、维布妥昔单抗(CD30 ADC)、贝林司他(HDAC)和米托蒽醌脂质体。国内获批的仅有西达本胺、普拉曲沙和戈利昔替尼。后续在研产品的靶点集中在:PI3K、HDAC、EZH2、JAK等。
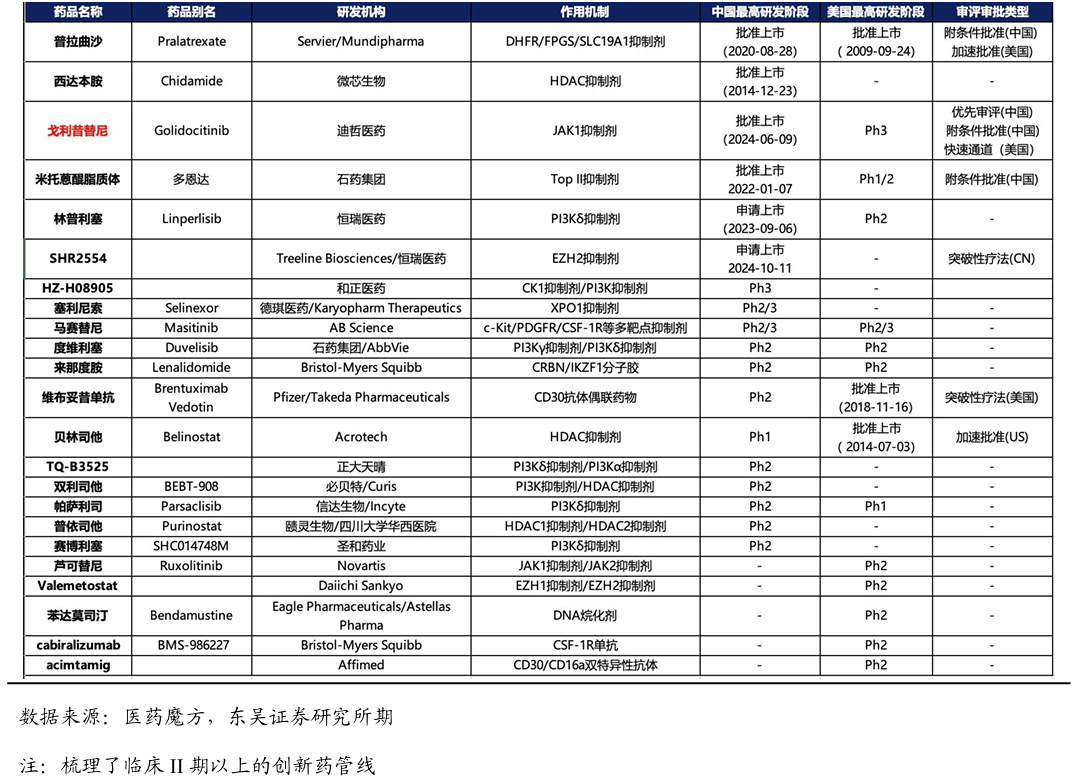
r/r PTCL从有效性角度看,戈利昔替尼带来更深缓解、更长生存。目前,针对r/r PTCL的疗效确切,明显优于现有疗法,戈利昔替尼的ORR=44.3%,CR=24%,mPFS=5.6个月,mDOR=20.7个月。入组患者的基线较差,超过73%的患者经过2线及以上治疗,50%患者经过HDAC抑制剂治疗,10%患者经过CD30靶向治疗。值得注意的是,在既往接受HDAC抑制剂、CD30靶向治疗以及米托蒽醌的患者中也显示出了疗效,体现了戈利昔替尼的潜在优效性。非头对头临床数据显示:mDOR在10个月左右,只有维布妥昔单抗联合化疗的mDOR接近20个月,而戈利昔替尼单药超过了20个月。林普利塞的mPFS较高,但是入组患者的基线相对较好。
r/r PTCL从安全性角度看,戈利昔替尼的安全性优势明显。JACKPOT8 临床数据显示:戈利昔替尼组患者的三级以上的不良反应发生率为60%,没有发生患者SAE或者死亡,停药的患者占7%,减量的患者占8%。PI3K抑制剂、HDAC抑制剂和CD30 ADC产品的不良反应较大,存在的安全性风险较高。相比之下,戈利昔替尼的安全性优势明显,不良反应主要表现为血液学相关毒性。
从用药便捷度看,每日口服一次,患者依从性更高。维布妥昔单抗需要联合化疗需要去医院静脉注射,比较麻烦。相比之下,戈利昔替尼单药每日口服一次,更方便,患者依从性更好。
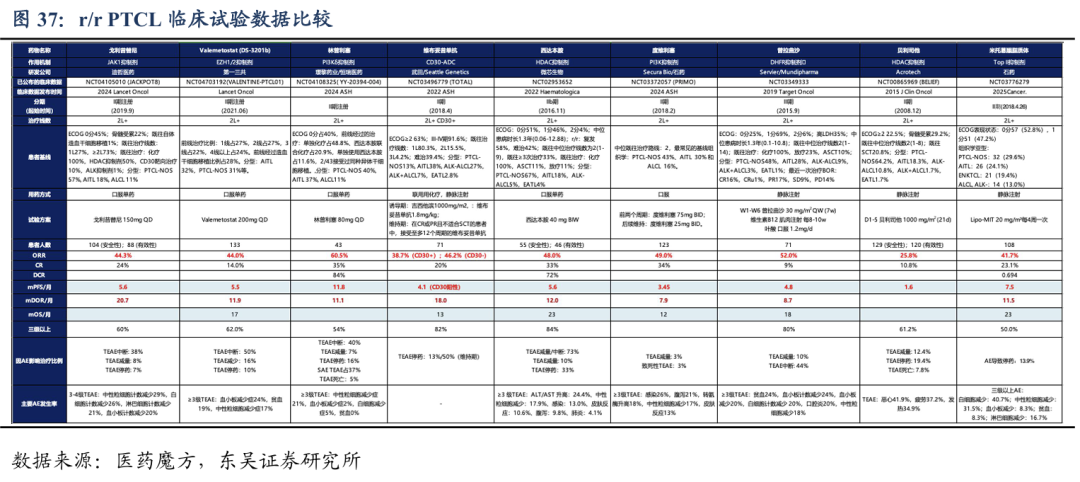
从治疗费用看,价格合适,医保后销售有望放量:戈利昔替尼2024年6月上市后,2024年底成功通过国家医保谈判,进入国家医保目录,2025年1月开始执行医保价格。12个月的治疗费用为18.7万元(按医保价格算)。相比国内没有进入医保的普拉曲沙用药费用优势明显,略高于西达本胺的年治疗费用。戈利昔替尼的化合物专利我们预计2036年到期,目前还没有普拉曲沙、西达本胺和米托蒽醌脂质体的仿制药上市。我们预计最近几年戈利昔替尼的销售不会受到仿制药的冲击。
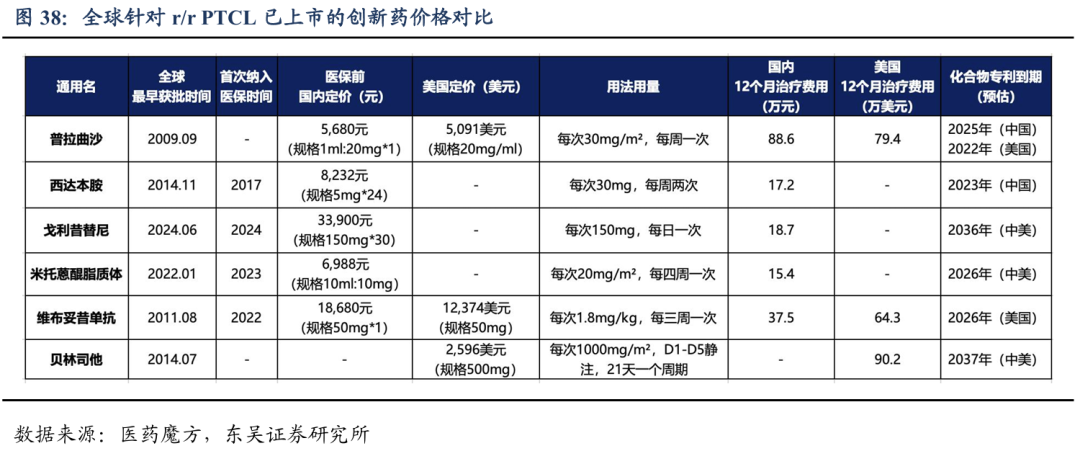
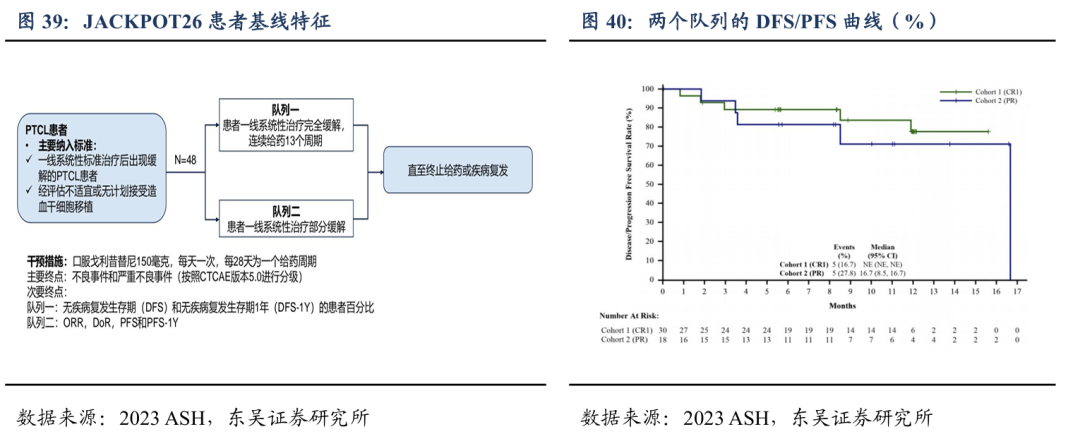
戈利昔替尼对于PTCL患者一线系统性治疗后的维持/巩固治疗效果显著。PTCL患者在接受一线标准治疗后,约40%的CR患者和80%的PR患者在初次肿瘤缓解后的2年内会出现疾病复发或进展,患者预后极差,缺乏标准的维持治疗。临床II期(JACKPOT26)的研究结果表明:患者一线系统性治疗CR的患者(队列1),中位随访12个月,76.7%的患者未观察到DFS事件(疾病进展);一线系统治疗PR的患者(队列2),中位随访时间10个月,mPFS=16.7个月,61%患者仍没有发生疾病进展。最常见的3级及以上的TEAE为血液学毒性,大多数可恢复或临床可管理且耐受性良好。
3.4.戈利昔替尼在自身免疫性疾病中差异化探索
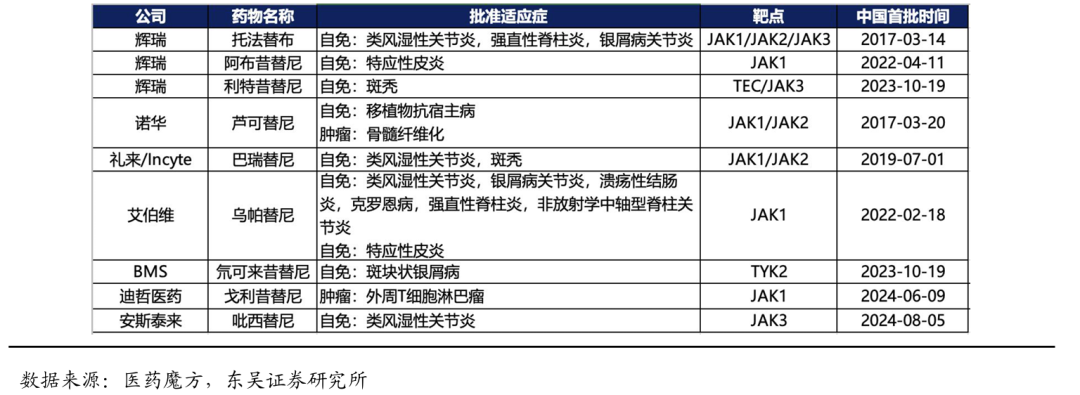
截至2025年1月,中国已批准上市的JAK 抑制剂共9款,除了诺华的芦可替尼获批骨髓纤维化以及迪哲的戈利昔替尼获批PTCL肿瘤适应症,其他JAK抑制剂主要获批适应症为类风湿性关节炎、银屑病关节炎等自身免疫性疾病。JAK抑制剂在自身免疫性疾病中的潜力较大,包括:白癜风、斑秃、炎症性肠病等,在研的品种个数繁多。
2023年11月,迪哲医药与无锡市高发投资发展集团有限公司(简称“高发集团”)共同出资设立合资公司,其中,迪哲医药出资7亿元,持股比例为87.5%;高发集团出资1亿元,持股比例为12.50%。在特应性皮炎、白癜风、慢性自发性荨麻疹和斑秃的局部治疗领域进行戈利昔替尼以及DZD8586的药品的研发、生产及销售。
3.5.戈利昔替尼的销售峰值测算
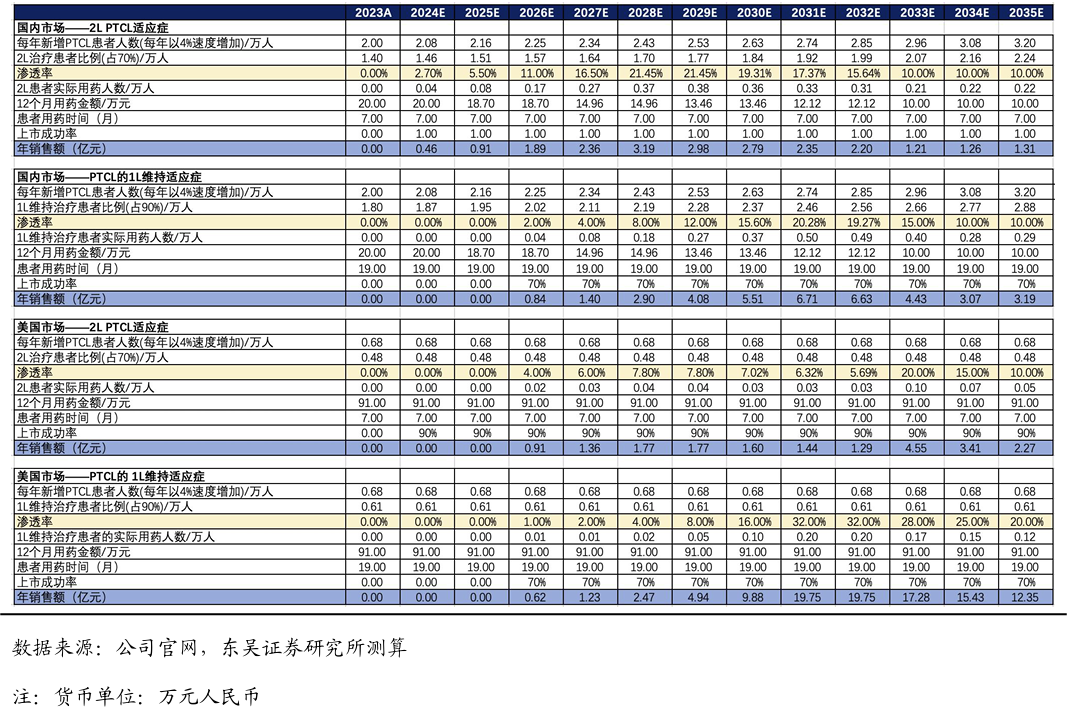
上市时间假设:戈利昔替尼已于2024年6月国内获批上市,我们预计2026年年底美国获批上市,欧盟、英国等海外国家我们预计2026年年底陆续获批上市。
药物降价幅度假设:戈利昔替尼2025年首次执行医保价格,国内12个月的治疗费用约为18.7万元,之后我们预计每两年降价一次,终局价格维持在8万元/年。美国的治疗费用我们预计达到91万元/年。
渗透率假设:针对r/r PTCL,戈利昔替尼的竞争格局良好,潜在同类最佳分子。假设二线及以上治疗峰值渗透率为21%;一线维持治疗的峰值渗透率有望达到20%。美国的渗透率有望分别达到20%和32%左右。
基于上述假设,舒沃替尼国内销售峰值我们预计达到10亿元,海外销售峰值我们预计达到6亿美金(美国4亿美金)。
4. DZD8586全球首创,克服BTK耐药突变
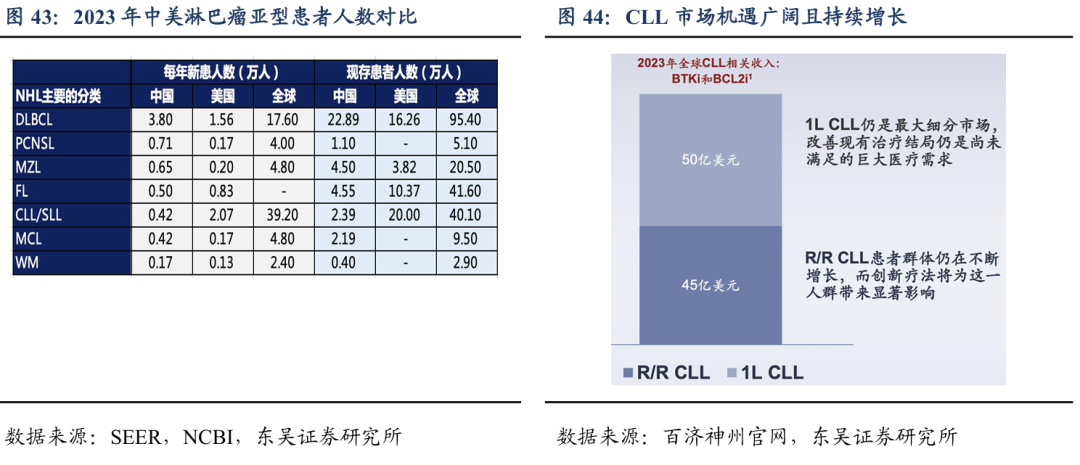
根据SEER和中国肿瘤诊疗指南,中国发病率最高的非霍奇金淋巴瘤(NHL)是DLBCL,每年新增3.8万人的DLBCL患者,存量患者近23万人;美国发病率第一的NHL是CLL/SLL,预计美国每年新增2万人CLL/SLL,存量患者约20万人。根据IQVIA分析,2023年全球CLL/SLL的市场有95亿美金,未来市场空间有望达到120亿美金。
DZD8586是公司自主研发且全球首创的一款针对B细胞非霍奇金淋巴瘤(B-NHL)的非共价LYN/BTK双靶点抑制剂,可完全穿透血脑屏障。目前市场上BTK抑制剂对部分B-NHL(如CLL和MCL)疗效显著,但耐药问题不可避免。并且,目前尚无BTK抑制剂被证明治疗DLBCL可取得显著临床获益。DZD8586有望解决BTK抑制剂的耐药问题并在DLBCL上取得疗效突破。
4.1. BTKi耐药后B-NHL仍存在较大未被满足的临床需求
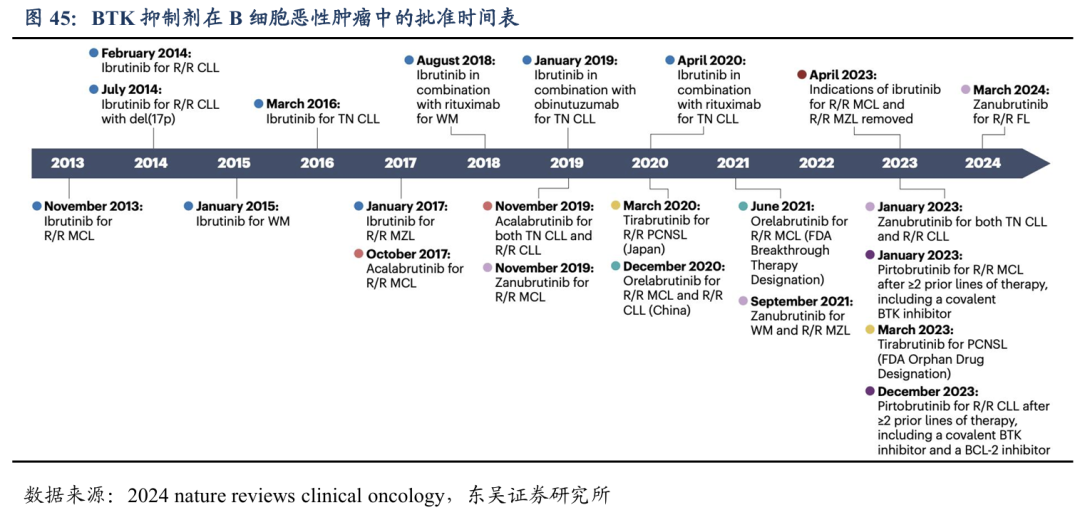
目前全球累计获批6款BTK抑制剂(BTKi),第一代 BTKi:强生/艾伯维的伊布替尼(Ibrutinib);第二代 BTKi:阿斯利康的阿卡替尼(Acalabrutinib)、百济神州的泽布替尼(Zanubrutinib)、奥布替尼(Orelabrutinib,仅中国)和吉利德科学/小野制药的替拉鲁替尼(Tirabrutinib,仅日本);第三代 BTKi:礼来的吡托布鲁替尼(Pirtobrutinib)。尽管BTKi对多种B细胞恶性肿瘤有效,但长期使用BTKi治疗不可避免地会导致耐药性。甚至在DLBCL适应症中目前仍没有BTKi获批上市。
在接受BTK抑制剂治疗的B-NHL患者中,已发现主要两种耐药机制:1、BTK基因突变,尤其是C481X突变阻断共价抑制剂与BTK的结合位点(图43b),BTK下游的PLCG2基因突变也是常见的耐药机制之一;2、BTK酶失活或活性显著降低引起引发非BTK依赖性BCR信号传导途径激活(非BTK依赖性突变)。当前,尚无药物能同时应对这两种耐药机制。
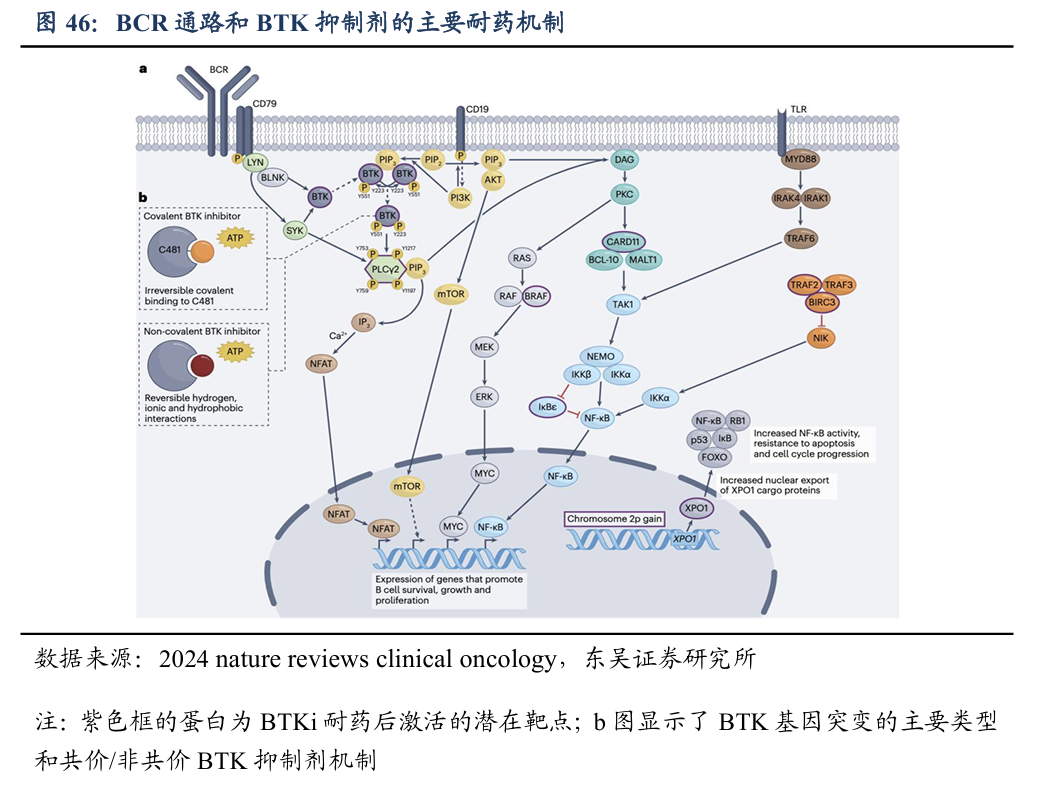
4.2全球首创LYN/BTK双靶点非共价抑制剂
DZD8586(非共价LYN/BTK双靶点抑制剂)可完全穿透血脑屏障,覆盖B-NHL全亚型。可同时阻断BTK依赖性和非依赖性BCR信号通路,有效抑制多种B-NHL亚型细胞生长且完全穿透血脑屏障。公司在全球均布局了化合物专利,专利有效期我们预计2040年到期。针对临床前实验显示DZD8586在DLBCL和CLL模型中显示出已上市的BTK抑制剂(阿卡替尼和吡托布鲁替尼)更强的抗肿瘤效果。
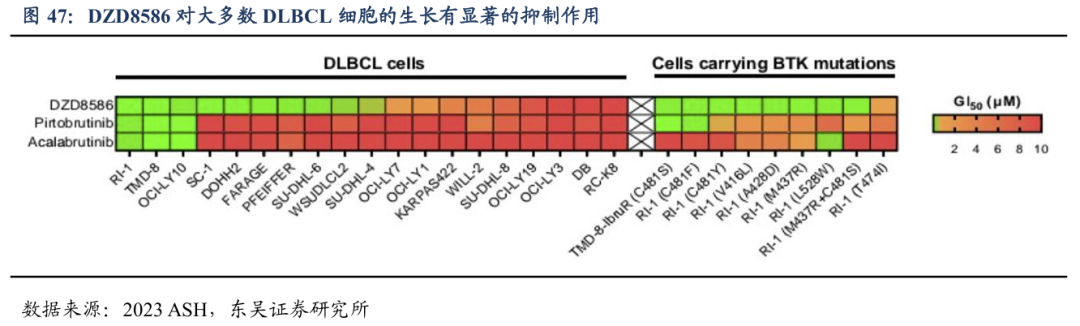
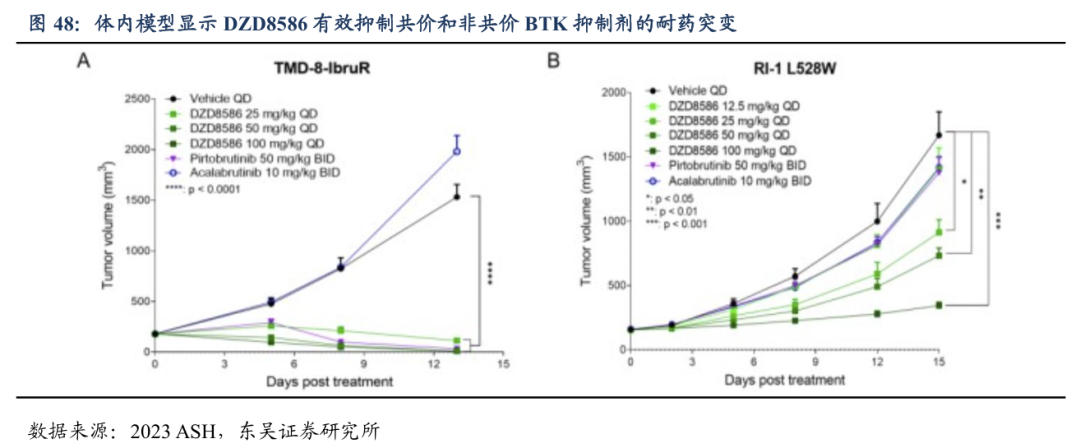
4.2. 1DZD8586在r/r CLL中疗效显著,安全性优异
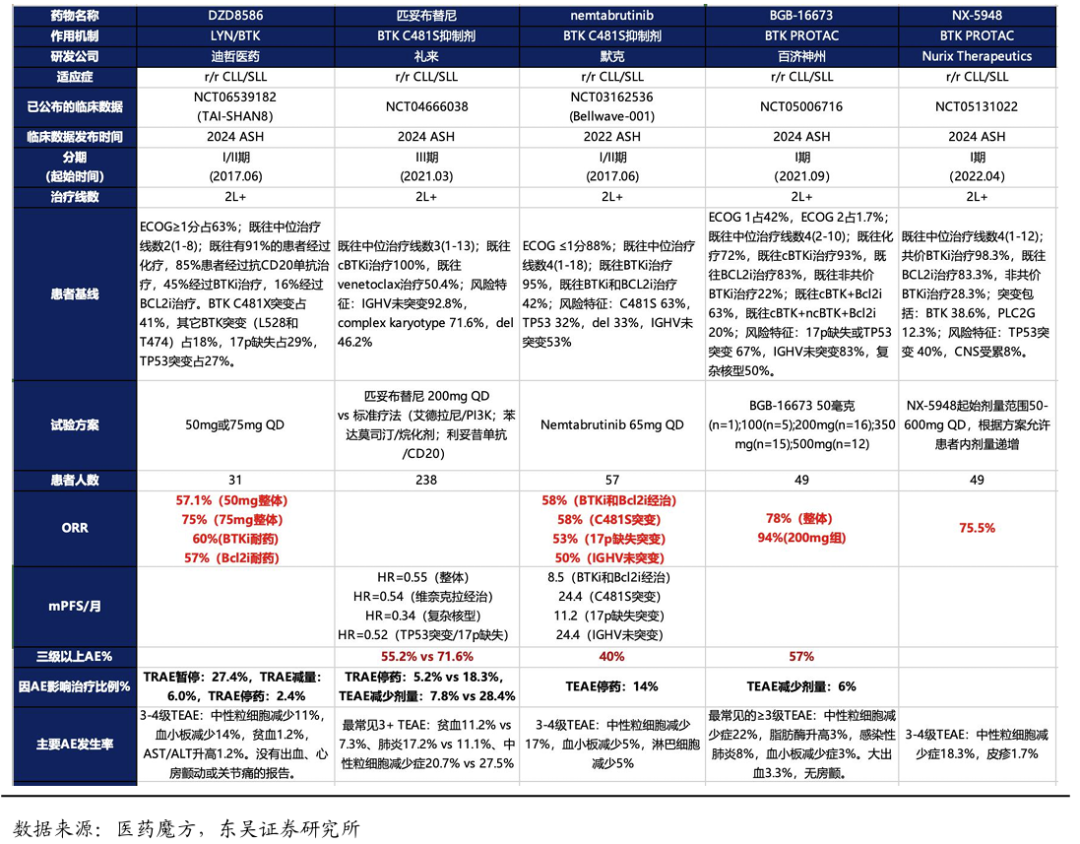
目前,1L CLL/SLL的患者仍以使用BTK抑制剂单药为主,耐药后可选择不同种BTKi再进行尝试,比如:可使用第三代BTK抑制剂吡托布鲁替尼,或者使用Bcl2抑制剂(维奈克拉)等,少数可以选择免疫疗法,包括奥妥珠单抗、利妥昔单抗联合疗法等。在研的BTK降解剂(PROTAC或CDAC)也在临床试验进行中,只有百济神州的BGB-16673读出了Ph1/2临床数据。
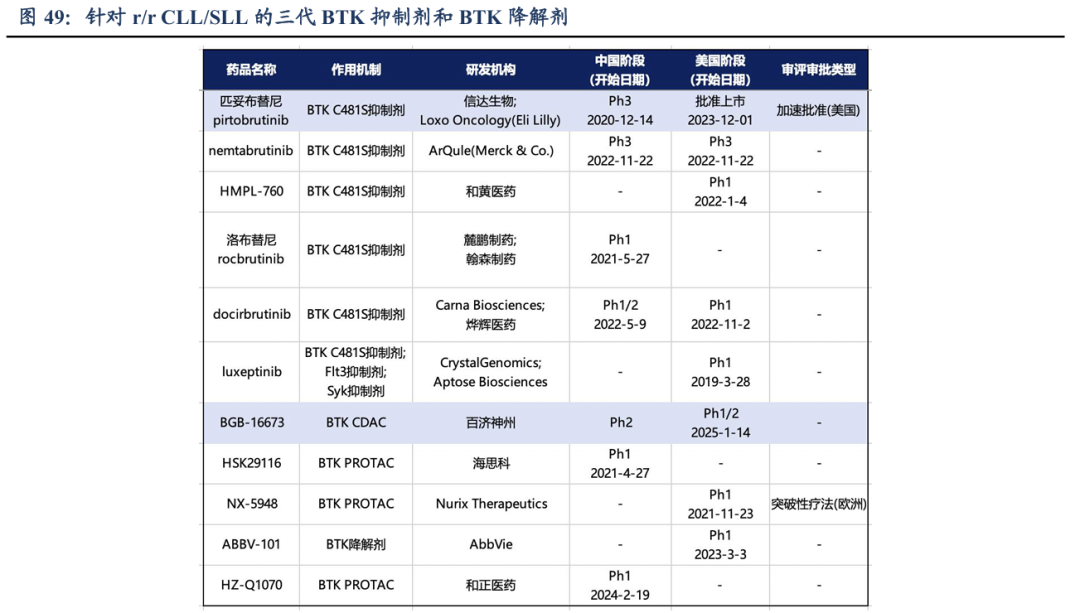
目前,礼来的非共价BTK抑制剂(ncBTKi)吡托布鲁替尼已经进入临床III期,患者基线都为共价BTK抑制剂(cBTKi)耐药的患者,其中50%的患者是Bcl2抑制剂维奈克拉耐药的,相比标准疗法mPFS获益明显,mPFS的风险获益比(HR)都在0.55以下。
百济神州的BTK CDAC公布的早期数据显示,中位治疗线数平均为4的患者中,既往经过ncBTKi治疗的占比为22%;既往cBTK+Bcl2i 占63%,既往cBTK+ncBTK+Bcl2i占 20%,患者基线水平较差,ORR仍高达78-94%。但是相比小分子抑制剂安全性不够好,57%的三级以上不良反应发生率,大出血发生率3.3%,治疗期间的停药率达14%。
2024年底的ASH会议上,DZD8586针对r/r CLL/SLL 的II期研究结果(TAI-SHAN8,数据截至2024年10月20日)显示:中位治疗线数为2,既往有91%的患者经过化疗,85%患者经过抗CD20单抗治疗,45%经过BTKi治疗,16%经过BCL2i治疗。在50mg和75mg剂量下的ORR分别为57.1%和75%。在既往接受过BTKi和BCL2i的患者中ORR分别为60%和57%。在携带经典BTKi耐药的(C481X突变)和非BTK依赖性突变中均观察到肿瘤缓解。安全性良好,药物相关导致的剂量暂停、减量和停药的发生率分别为27.4%、6.0%和2.4%,主要为血液学毒性,无药物相关死亡。
综上,相比吡托布鲁替尼,DZD8586的有效性潜在更高;相比百济新一代的BTK CDAC安全性有潜在优势。更重要的是,无论是ncBTKi还是BTK降解剂都只能针对BTK突变的患者,DZD8586作为双靶点抑制剂,有望解决非BTK依赖性突变的耐药问题。

4.3. DZD858有望突破DLBCL治疗范式,提供更全面的抗肿瘤效应
近20年来,R-CHOP(利妥昔单抗联合环磷酰胺、多柔比星、长春新碱和泼尼松)6-8个疗程一直是弥漫大B细胞淋巴瘤(DLBCL)患者的标准一线治疗方案,R-CHOP方案的出现显著提升了DLBCL患者一线的治愈率,5年PFS率与OS率攀升至约60%与约77%的水平,仍有30%患者复发或难治。后续不断有研究者希望通过调整R-CHOP剂量或增加小分子抑制剂比如硼替佐米或伊布替尼等来替代R-CHOP方案,但大多都以失败告终。DLBCL具有很强的异质性,目前RCHOP方案可以治愈60%~70%患者,但是近年来新的靶向小分子、抗体药物偶联物(ADC)、双特异性抗体、CAR-T等新分子实体在DLBCL领域不断探索,有望创造出能够超越R-CHOP的新治疗方案以及解决r/r DLBCL疗效不佳的困境。DZD8586目前针对DLBCL正在国内进行临床I/II期,有望在2025年更新数据。
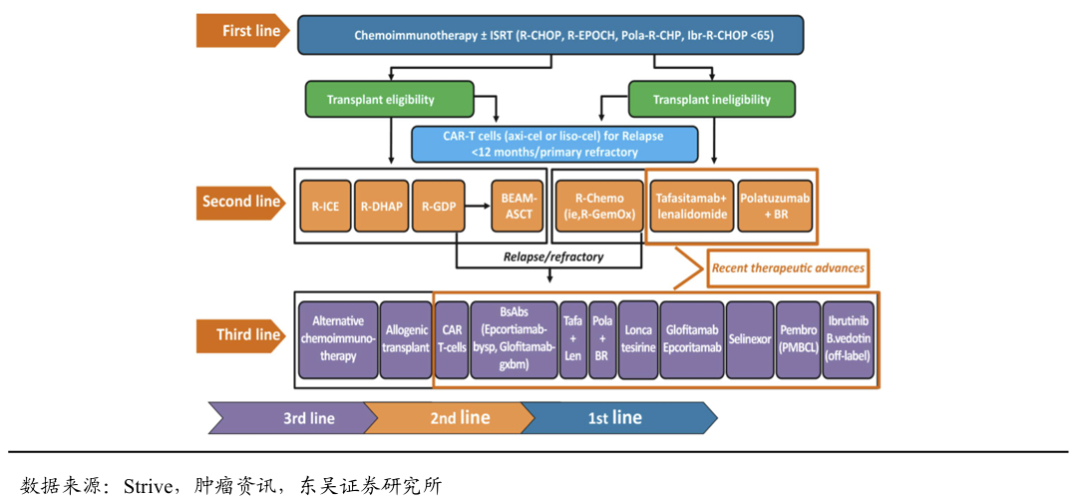
4.4. DZD8586的销售峰值测算
上市时间假设:DZD8586针对r/r CLL/SLL和r/r DLBCL适应症的国内注册临床有望2025年启动,我们预计于2028年国内获批上市,暂不考虑海外市场。
药物降价幅度假设: DZD8586 我们预计上市首年国内12个月的治疗费用约为22万元,之后我们预计每两年降价一次,终局价格约8万元/年。
渗透率假设:针对r/r CLL/SLL和r/r DLBCL适应症,DZD8586的竞争格局良好,潜在同类最佳分子。假设国内峰值渗透率为22%。
基于上述假设,DZD8586国内销售峰值我们预计达到10亿元,海外市场暂不计入销售测算。
5. DZD6008全球首创,攻克肺癌耐药困境
5.1. EGFR TKI治疗耐药的EGFRm NSCLC患者仍缺乏治疗药物
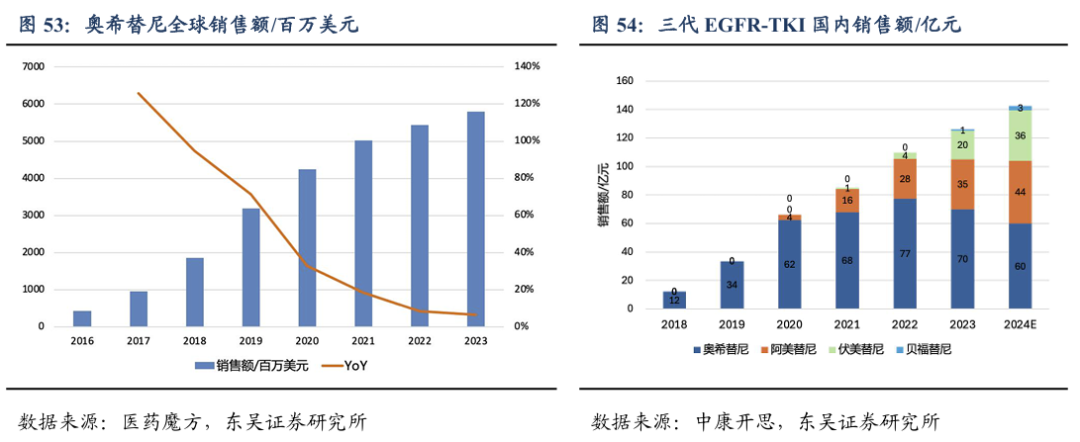
第三代EGFR TKI目前已成为1L EGFRm NSCLC治疗的主流,代表药物为奥希替尼在2023年全球总销售额近60亿美元,我国三代EGFR TKI市场我们预计会达到150-200亿人民币。但与一代和二代EGFR-TKI类似,接受三代TKI治疗的患者不可避免地会出现获得性耐药,且目前已发现多种耐药机制。EGFR TKI治疗后耐药的EGFRm NSCLC市场空间仍然广阔。
三代EGFR TKI耐药机制复杂,包括:脱靶耐药(Off-target)和靶点依赖性耐药(On-target)。三代 EGFR-TKI 耐药机制的类型大体分为以下三种:
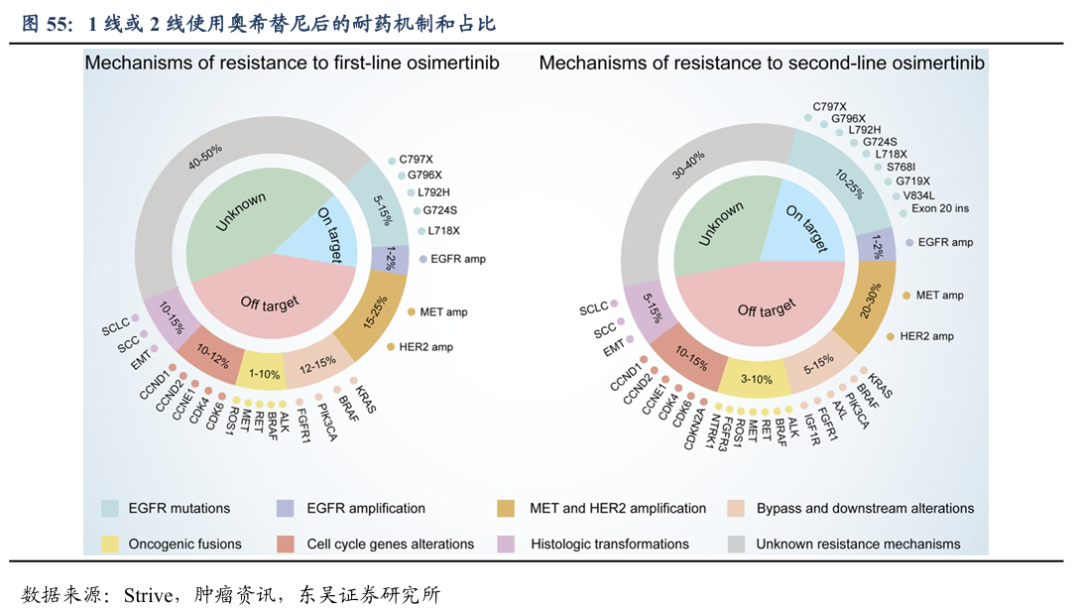
EGFR依赖性耐药:阻止EGFR-TKI抑制靶受体酪氨酸激酶的突变,点突变约占一线使用奥希替尼耐药后的5%-15%。C797S是最常见的EGFR依赖性耐药点突变之一,一、二线奥希替尼耐药后发生C797S突变的比例分别为7%和11%。
旁路或下游激活:尽管有足够的TKI结合,但引起旁路和/或下游信号通路的激活,促进细胞存活和增殖。一、二线奥希替尼耐药后发生MET扩增和HER2扩增的比例分别为15%-25%和20-30%。
组织学或表型转化:如从腺癌转变为鳞状细胞癌或小细胞肺癌癌症表型和上皮-间质转化(EMT)。一、二线奥希替尼耐药后发组织学转化的比例分别为10%-15%和5-15%。
5.2. DZD6008全球首创,临床推进快速开展
DZD6008是迪哲自主研发的、全新的针对NSCLC的小分子抑制剂,有望填补当前未被满足的临床空白,正在中国开展针对TKI治疗失败的EGFRm NSCLC的I期临床研究。有望今年在学术会议上更新早期临床结果。尤其是TKI耐药后的NCLCL的脑转移是导致患者死亡的主要原因,近30% EGFRm NSCLC患者在初次诊断时即存在脑转移,诊断后3年内脑转移的风险可能增至50-60%。目前包括ADC、双抗在内大分子药物穿透血脑屏障方面存在一定的局限性。

5.3. TKI耐药后EGFRm NSCLC市场广大,在研疗法百花齐放
尽管在第三代EGFR-TKI耐药后可以通过再次活检来确定耐药机制,但仍有高达 30%-50%的患者耐药机制不明。针对这类患者,传统疗法是基于铂类的姑息性化疗,ORR仅25%,PFS仅4.3个月,获益有限。第四代EGFR TKI主要针对C797X突变,还未有药物获批上市,研发难度较大。因此,临床上还进行了诸多分子实体的探索,并初步取得了积极的结果:
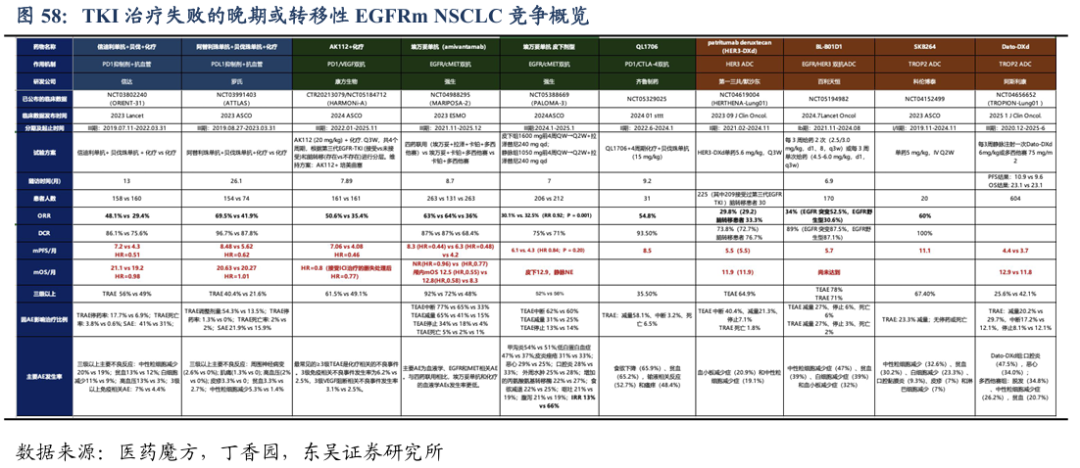
免疫联合化疗及抗血管治疗:1)阿替利珠单抗联合贝伐珠单抗和化疗的三药联合方案治疗EGFRm的NSCLC患者的III期结果(ATTLAS研究)。相比化疗PFS有改善(HR = 0.62,P = 0.004),但两组OS差异无统计学意义。另外,三药联合疗法组不良事件发生率更高;2)信迪利单抗+贝伐珠单抗+化疗对照化疗的III期临床结果(ORIENT-31研究)显示:PFS获得显著改善(7.2 个月 vs 4.3个月,HR=0.51)。生存获益HR为0.98。基于该研究结果,2023年国内批准了信迪利单抗联合贝伐珠单抗治疗经EGFR-TKI治疗失败的EGFRm的局部晚期或转移性非鳞状NSCLC。
双抗:1)III 期 MARIPOSA-2 研究,探索了埃万妥单抗联合化疗±拉泽替尼治疗(ACP- L/ACP方案)奥希替尼治疗后疾病进展的EGFRm晚期NSCLC患者的疗效和安全性。结果显示:与单纯化疗相比,ACP 和 ACP-L 方案分别降低了52%和56%的疾病进展或死亡风险,但 OS无显著差异;2)HARMONi-A研究是首个比较了依沃西单抗联合化疗与单独化疗在EGFR-TKI 耐药的 EGFRm晚期或转移性非鳞NSCLC疗效的III期研究。结果显示:依沃西单抗联合化疗组PFS获益显著,达到了主要分析终点(mPFS:7.06个月 vs. 4.8个月,HR = 0.46,p 0.001)。各亚组PFS获益情况与整体获益趋势一致。在OS方面,依沃西联合化疗OS曲线早期分离,有明显延长趋势。基于HARMONi-A研究,2024年5月,依沃西单抗正式国内获批上市。
抗体偶联药物(ADC):1)HER3-DXd是一种靶向HER3的ADC,治疗EGFRm局部晚期或转移性NSCLC患者的III期临床显示mPFS=5.5个月,mOS=11.9个月。2)III 期TROPION-Lung01研究评估了Dato-DXd 对照多西他赛在既往至少接受过一种治疗、伴或不伴驱动基因阳性的局晚或转移性NSCLC患者的有效性和安全性。结果显示,非鳞癌亚组mPFS具有显著获益,OS 有获益趋势。2024年2 月,基于TROPION-Lung01 研究,FDA 已受理了 Data-DXd 用于既往接受过标准治疗的晚期非鳞状NSCLC申报上市申请。3)芦康沙妥珠单抗(SKB264/sac-TMT)是一种靶向TROP-2 的ADC。KL264-01(MK2870-001)研究是一项芦康沙妥珠单抗针对NSCLC等重度经治晚期实体瘤患者开展的I/II 期临床试验。结果显示,EGFRm耐药患者ORR达60.0%,mDoR 为 9.3 个月,mPFS长达11.1个月。III 期SKB264-Ⅲ-09研究则探索了SKB264 单药对比培美曲塞+铂类用于EGFR-TKI 治疗失败的 EGFRm局晚期或转移性 NSCLC,该研究正在开展中。4)百利天恒的EGFR-HER3 ADC在早期研究中单药治疗EGFRm NSCLC的后线患者:mPFS=5.7个月。
5.4. DZD6008的销售峰值测算
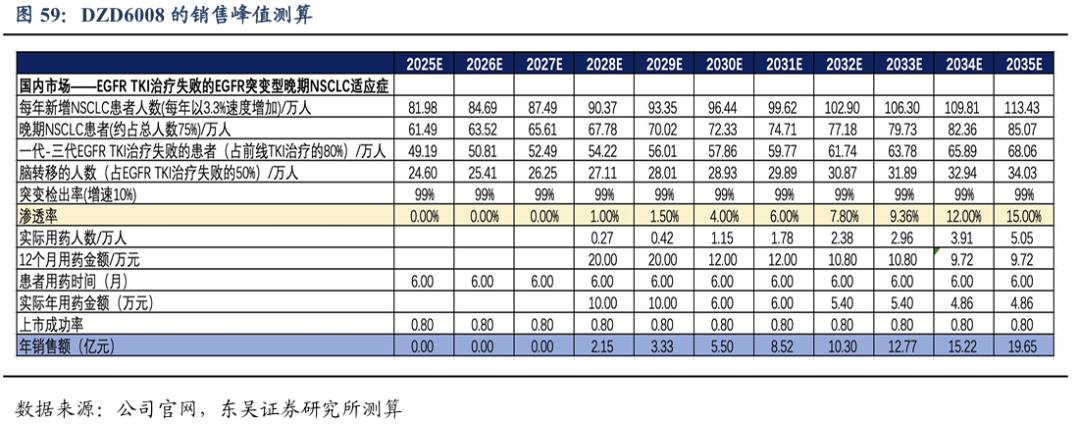
上市时间假设:DZD6008针对EGFR TKI耐药晚期、转移性EGFRm NSCLC适应症的国内注册临床有望2025年启动,我们预计于2028年国内获批上市,暂不考虑海外市场。
药物降价幅度假设: DZD6008 我们预计上市首年国内12个月的治疗费用约为20万元,之后我们预计每两年降价一次,终局价格约不到10万元/年。
渗透率假设:针对晚期、转移性EGFRm NSCLC适应症的竞争虽然激烈,但是DZD6008口服给药,透脑性好,小分子领域竞争格局良好,潜在同类最佳小分子。假设国内峰值渗透率为15%。
基于上述假设,DZD6008国内销售峰值我们预计达到20亿元,海外市场暂不计入销售测算。
6.盈利预测和估值
6.1.产品销售峰值预测
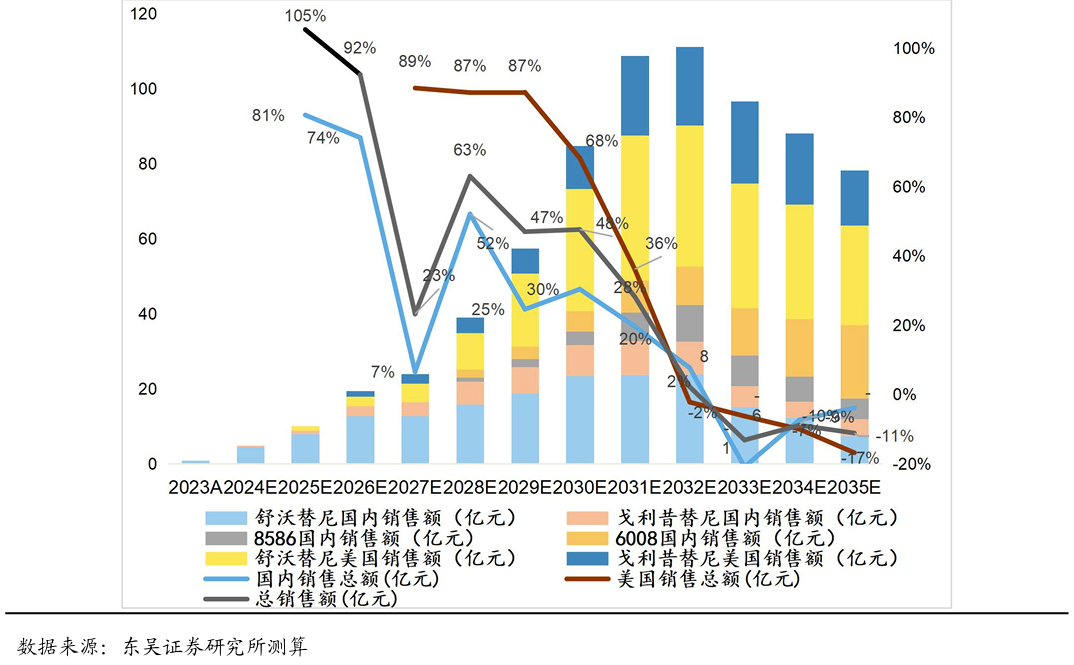
四个产品国内销售峰值我们预计达到近60亿元人民币。其中,舒沃替尼国内销售峰值为20亿元,戈利昔替尼国内销售峰值为10亿元,DZD8586国内销售峰值为10亿元,DZD6008国内销售峰值为20亿元。
舒沃替尼和戈利昔替尼海外销售峰值我们预计分别达到9亿美金和6亿美金。由于其它管线产品海外临床试验还未取得概念验证性结果,暂不考虑DZD8586和DZD6008的海外销售。
6.2.估值评级
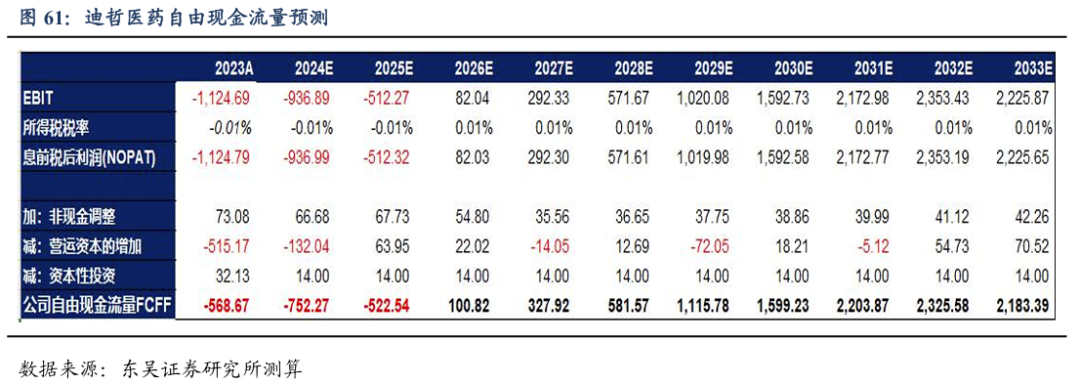
采用绝对估值法中的FCFF估值:迪哲医药研发能力强,临床推进快,目前已经实现了两款国内上市的创新药,有望今明两年海外实现两款创新药的上市。在研管线竞争格局良好,持续推进新产品在中国和海外实现获批上市,我们给1%的过渡期增长率和0.5%的永续增长率,贝塔值β为1,WACC值为5.17%。根据下表可以得出,公司的四款产品的绝对估值总数为343亿人民币,对应目标股价为82.26元/股。迪哲医药国内商业化顺利推进,产品出海确定性高,在研品种潜力大,首次覆盖给予“买入”评级。
公司目前仍处在未盈利状态,不适合传统的P/E相对估值方法。公司进入了商业化阶段,有产品的销售收入,但是国内销售还处在放量初期阶段,海外销售我们预计今年才开始,并且海外销售大多采取合作方分成的模式,因此我们选取了P/S和P/E结合的分步相对估值计算方式。我们选取了部分A股创新公司作为可比公司,包括:海思科、泽璟制药、艾力斯和奥赛康。可比公司2023年-2025年的P/S平均值为20、14和9.2。迪哲医药2025年的P/S为24.8高于可比公司的平均值9.2,主要原因是迪哲医药的产品处在放量初期,销售收入还在快速增长阶段。
我们预计迪哲的国内4个产品的销售收入我们预计2032年达峰,峰值为60亿元,按照9.2倍PS计算,折现后(10%折现率10%)贡献约257亿元市值;海外暂时只考虑舒沃替尼和戈利昔替尼的销售,销售峰值我们预计15亿美金(2031年),约贡献15亿人民币利润(行业平均的10%左右分成),按照行业的15倍PE计算,折现后(10%折现率10%)贡献约105亿元市值。综上,公司的目标市值(国内和海外)为362亿元。公司市值弹性较大,首次覆盖给予“买入”评级。

7.风险提示
新药研发及审批进展不及预期:公司多款创新药及新适应症研发处于临床I/II期阶段。而产品未来收入的增长主要来源于新产品和新适应症的获批,若临床试验或后续结果不及预期,公司将面临收入不及预期等风险。
药品的销售不及预期:价格降幅超预期的风险及市场格局竞争加剧的风险。
政策影响对产品价格的不确定性:考虑到医保谈判落地的影响,数量变化可能无法弥补价格降低带来的影响。
海外授权不及预期:如果海外合作没有如期达成,会影响产品海外市场的商业化销售。