投资要点
投资要点
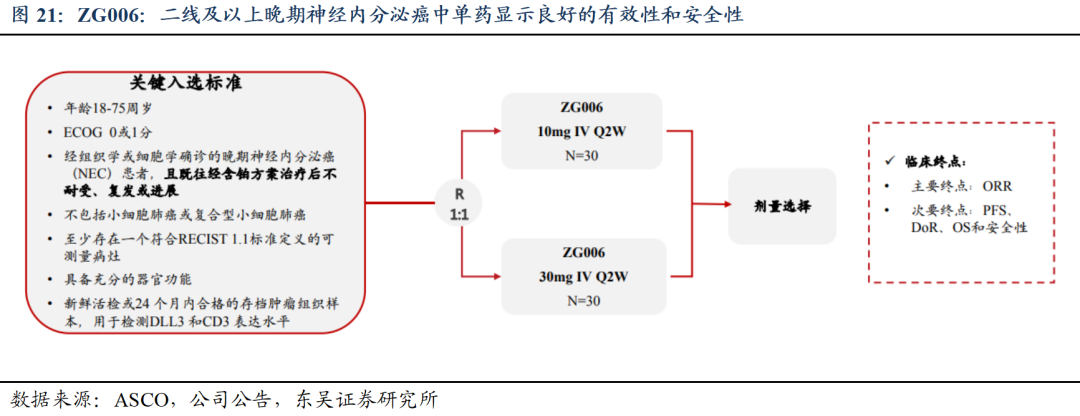
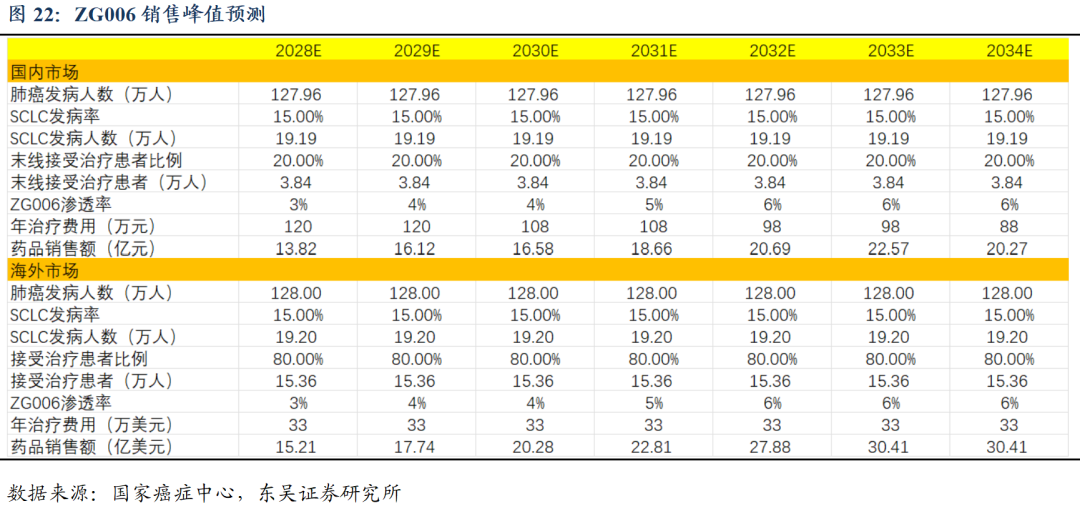
ZG006(CD3/DLL3/DLL3)疗效数据优异,打造小细胞肺癌基石疗法:ZG006为结构新颖的DLL3三抗,2025ASCO数据亮眼,针对三线及以上SCLCⅡ期剂量优化试验中,10mg与30mg组ORR分别为62.5%和58.3%,DCR分别为70.8%和66.7%。此外在神经内分泌癌患者中也展现显著抗肿瘤活性及良好的安全性。未来 ZG006针对后线SCLC及一线联合都将开展注册临床,全面布局小细胞肺癌赛道,打造基石疗法。海外临床获得FDA同意,有望近期入组患者。我们预计ZG006小细胞肺癌适应症国内销售峰值约20亿人民币、海外销售峰值约30亿美金,由于产品创新性强,初步数据优异,海外授权潜力较大。
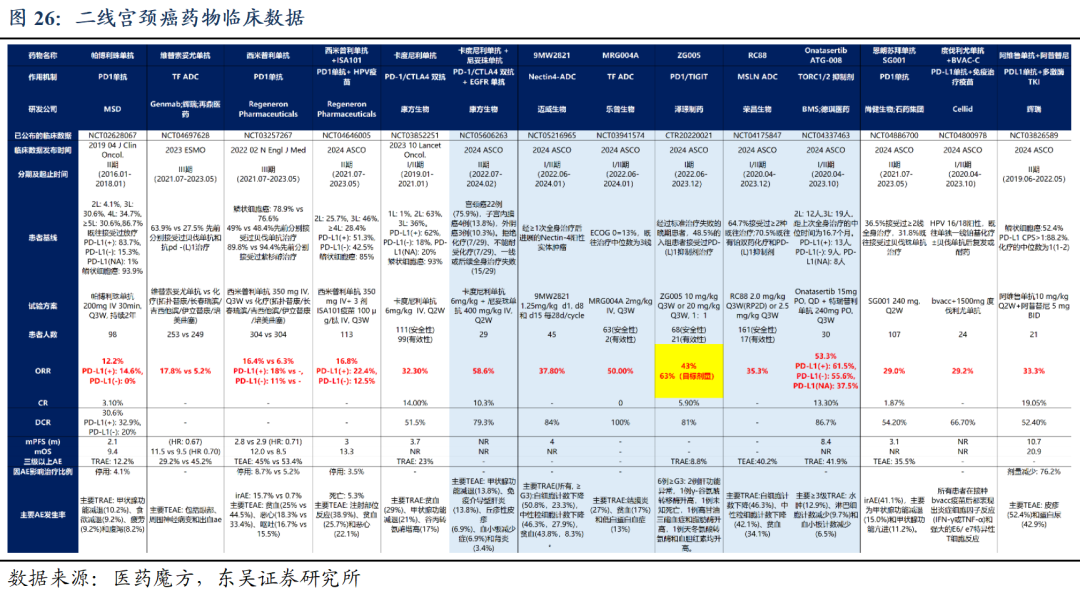
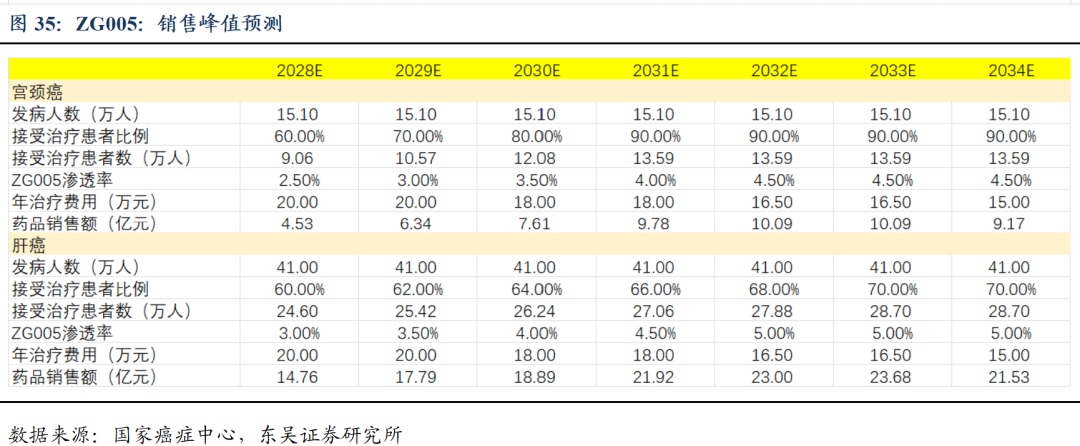
ZG005(PD-1/TIGIT)初步疗效已验证,泛瘤种潜力有望成为重磅炸弹产品: ZG005在既往未接受过免疫检查点抑制剂治疗的二线及以上宫颈癌患者中,20mg/kg组基于IRC评估的确认ORR为40.9%,DCR为68.2%。基于研究者评估,20mg/kg组mPFS已超过11个月。而在一线晚期宫颈癌患者中同样显示优异的有效性和安全性(20mg、10mg/kg组未确认ORR分为为82.1%和65.4%,DCR分别为96.4%和96.2%)。我们预计ZG005宫颈癌及肝癌适应症国内销售峰值约30亿人民币。此外,研究表明ZG005与化疗、ADC、TCE等治疗手段具备广泛联用潜力,且适应症拓展潜力较大,有望成为下一个重磅品种。
公司重磅产品陆续上市,进入商业化新阶段:吉卡昔替尼骨髓纤维化适应症成功获批上市,其最佳脾缓解率为80.9%,远高于芦可替尼,有望成为BIC药物;重组人凝血酶具备突出的临床止血效果及良好的安全性,已与远大生命达成独家商业化合作,未来有望快速放量;多纳非尼肝癌、RAIR-DTC两款适应症均纳入医保,销售额稳健增长;重组人促甲状腺激素(rhTSH)甲状腺癌术后诊断适应症已递交上市申请,研发进展处于国内前列,填补甲状腺诊疗空白,并与德国默克达成独家商业化合作,获得总授权2.5亿元。随着重磅产品陆续上市,公司销售收入有望大幅增长,贡献稳定现金流。
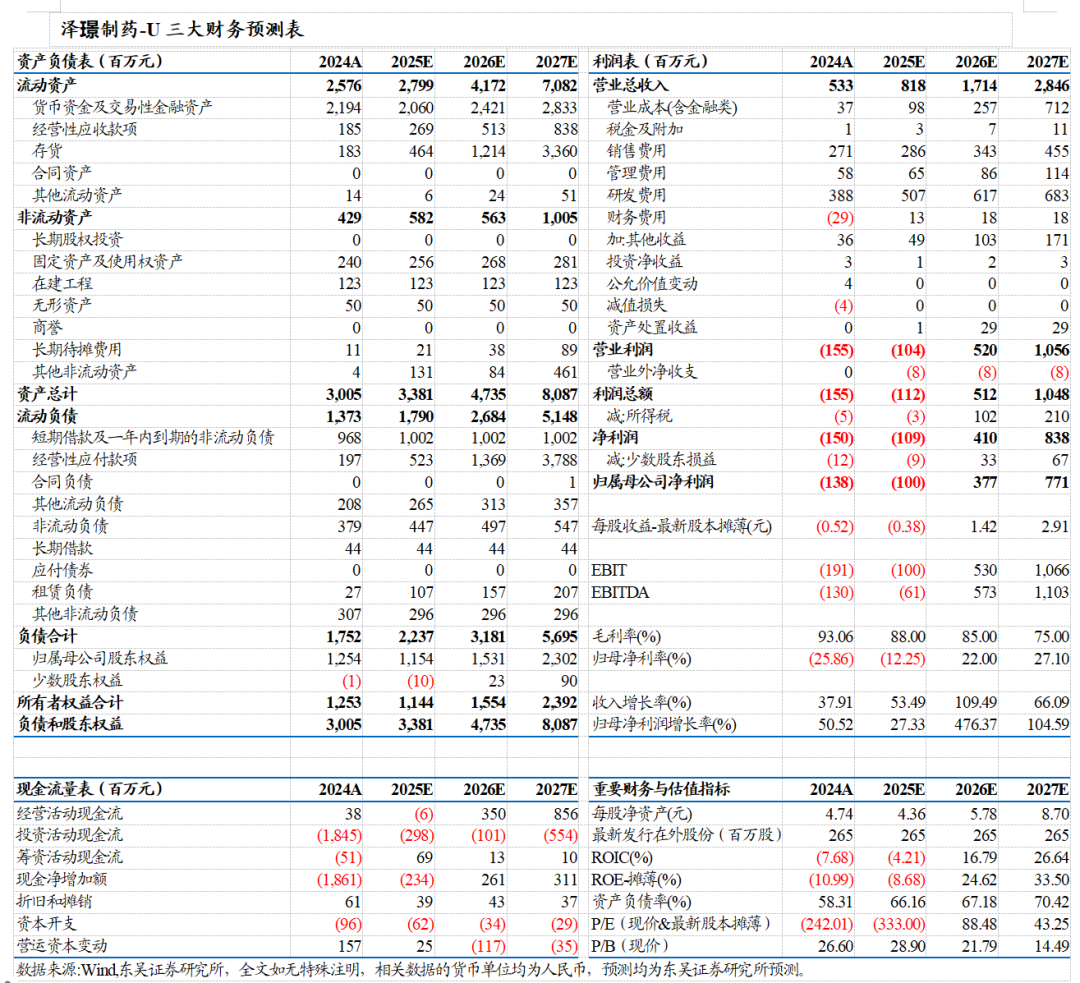
盈利预测与投资评级:公司多款产品成功商业化,在研管线层次丰富,已实现从Biotech向Biopharma转型。预计公司已商业化产品(多纳非尼、吉卡昔替尼、重组人凝血酶)合计销售峰值50亿元(给予3倍PS),在研核心产品(rhTSH、ZG006、ZG005)国内销售峰值约60亿元(给予3倍PS),ZG006海外销售峰值约30亿美金(考虑BD后销售分成给予10倍PE),可支撑公司市值530亿元。我们预计公司2025-2027年收入分别为8.18/17.14/28.46亿元,对应当前市值的PS为41/19/12倍,首次覆盖,给予“买入”评级。
风险提示:新药研发进展不及预期风险、药品销售不及预期风险、海外交易不及预期风险。



1.创新兑现:从Biotech到Biopharma
1.1 专注肿瘤、出血及血液治疗领域,高管及研发团队专业背景深厚
公司成立于2009年3月,致力于创新药物的自主研发、生产和商业化,并已成功建立了两个特色核心技术平台,即小分子药物研发及产业化平台、复杂重组蛋白新药和抗体新药研发及产业化平台。依托这两个技术平台,公司开发了丰富的小分子新药与重组蛋白新药的产品管线,覆盖肝癌、非小细胞肺癌、结直肠癌、甲状腺癌、鼻咽癌、骨髓增殖性肿瘤等多种癌症和血液肿瘤,出血、免疫炎症性疾病等多个治疗领域。
高管及研发团队专业背景深厚,在新药研发领域经验丰富。公司创始人、董事长兼总经理盛泽林于美国迈阿密大学取得药理学博士学位,曾任职于美国施贵宝公司、上海赛金生物、上海奥纳医药、白鹭医药等多家药企。此外公司多名核心技术人员均具有海内外知名药企的新药研发经验,专业背景深厚。

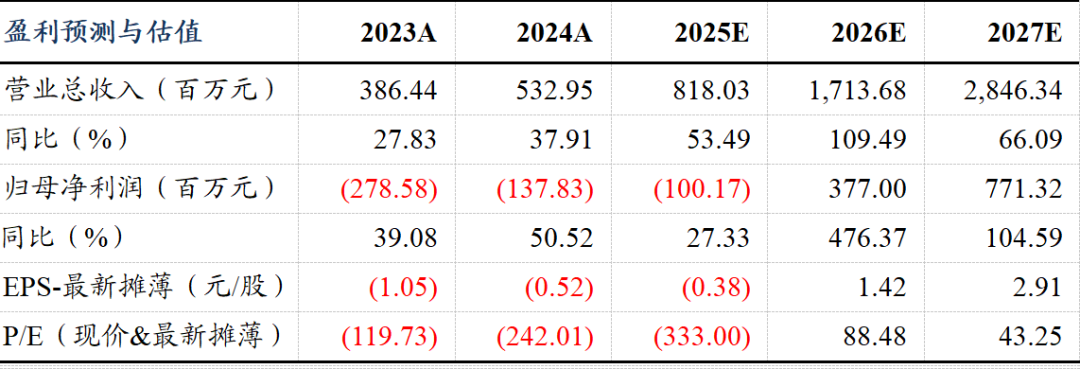
股权结构清晰,实控人为盛泽林及陆惠萍。截至2025年一季度末,两人分别持有公司股权18.85%、4.77%
1.2 创新收获已至,扭亏为盈在即
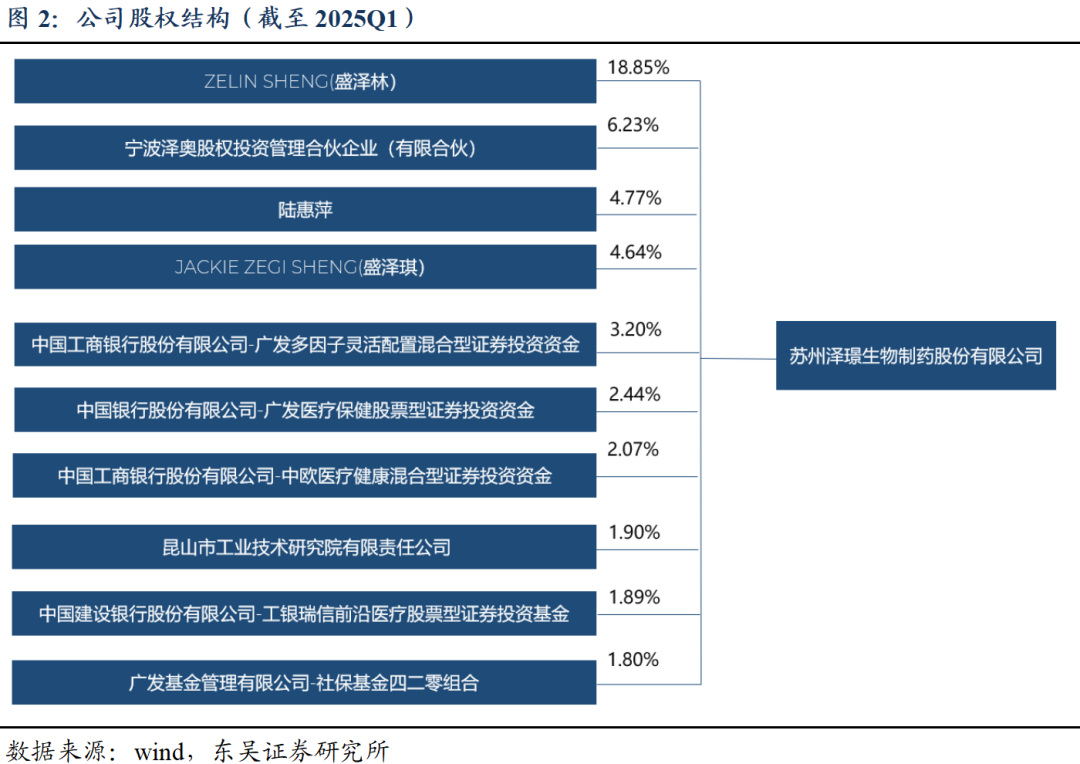
商业化兑现在即,公司有望扭亏为盈。公司2024年实现收入5.33亿元,同比增长37.91%;2025Q1实现收入1.68亿元,同比增长54.87%。自2022年以来,公司亏损逐年收窄,2024全年归母净利润亏损-1.38亿元,同比减亏1.40亿元,2025Q1归母净利润亏损0.28亿元。我们认为,随着公司近年来创新成果持续转化,获批上市产品如多纳非尼、吉卡昔替尼、重组人凝血酶纳入医保后预计快速放量,公司有望实现盈亏平衡、扭亏为盈。
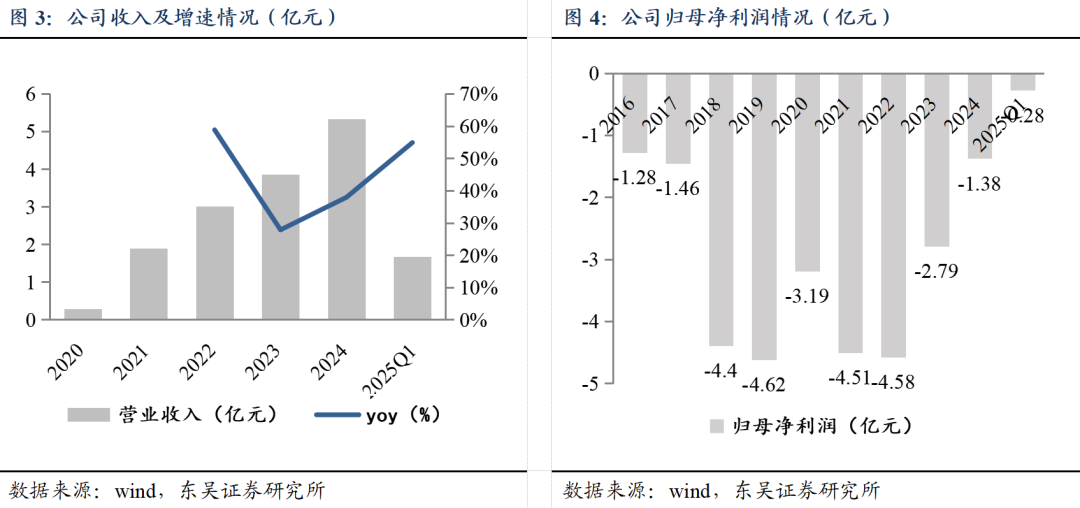
经营性净现金流转正。公司经营性现金流自2021年起逐步恢复,2024年因收到重组人凝血酶商业化合作首付款和里程碑款3.4亿元(2024年内收到2.8亿元),全年经营性净现金流由负转正。
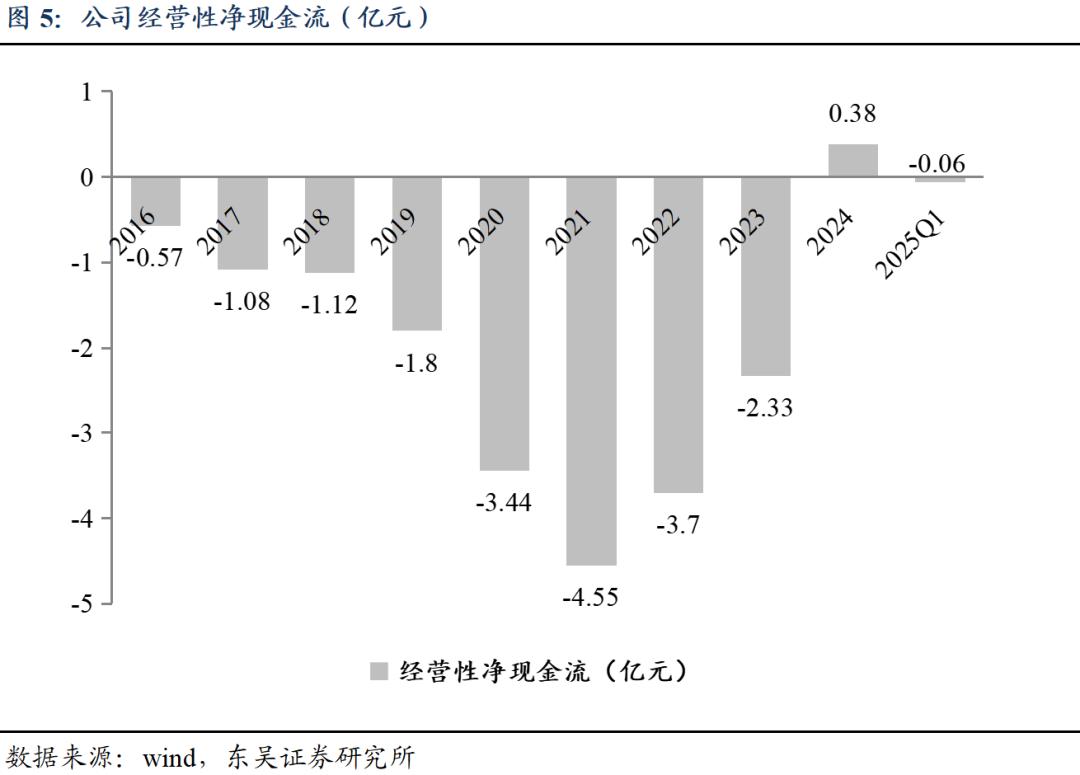
研发端持续投入,创新加码助力公司长久发展
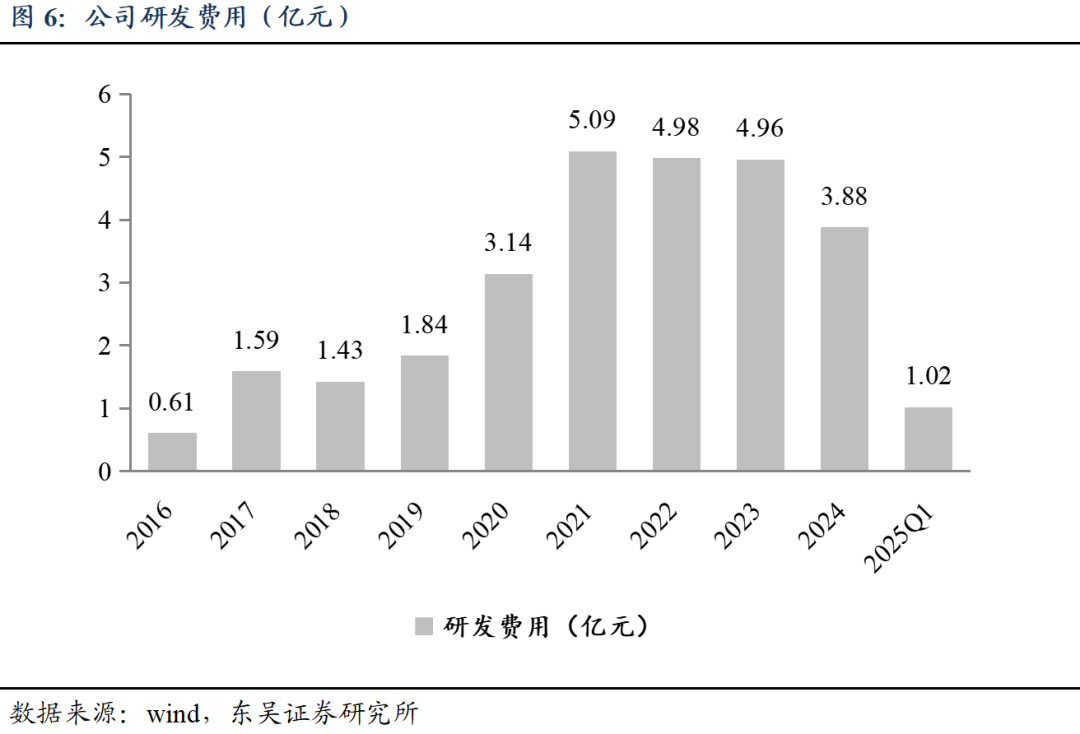
公司研发费用由2016年的0.61亿元增长至2023年的4.96亿元,2024年降低至3.88亿元,原因系具体研发项目所处不同研发阶段,整体研发费用同比有所减少。预计未来随着研发管线持续推进,公司将持续投入研发费用,加速创新兑现。
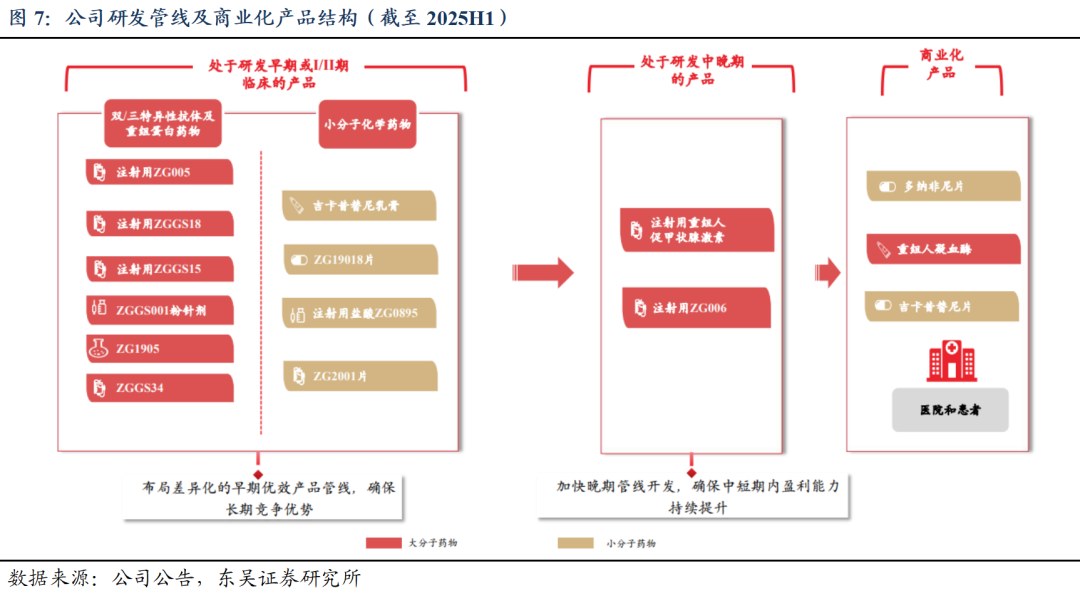
差异化管线布局、层次性推进研发进展,保障商业化优势及未来持续竞争力。公司在研管线丰富,截至2025H1,共计15项主要在研药品,涵盖双抗、三抗、重组蛋白药物及小分子化药。其中3款已获批上市产品(甲苯磺酸多纳非尼片晚期肝癌/RAIR-DTC、重组人凝血酶、盐酸吉卡昔替尼片MF)、1款BLA产品(rhTSH甲状腺癌术后诊断);2款产品的4项适应症处于Ⅲ期临床(盐酸吉卡昔替尼3项自免适应症、rhTSH甲状腺癌术后治疗);多款产品处于Ⅰ期或Ⅱ期临床试验阶段。
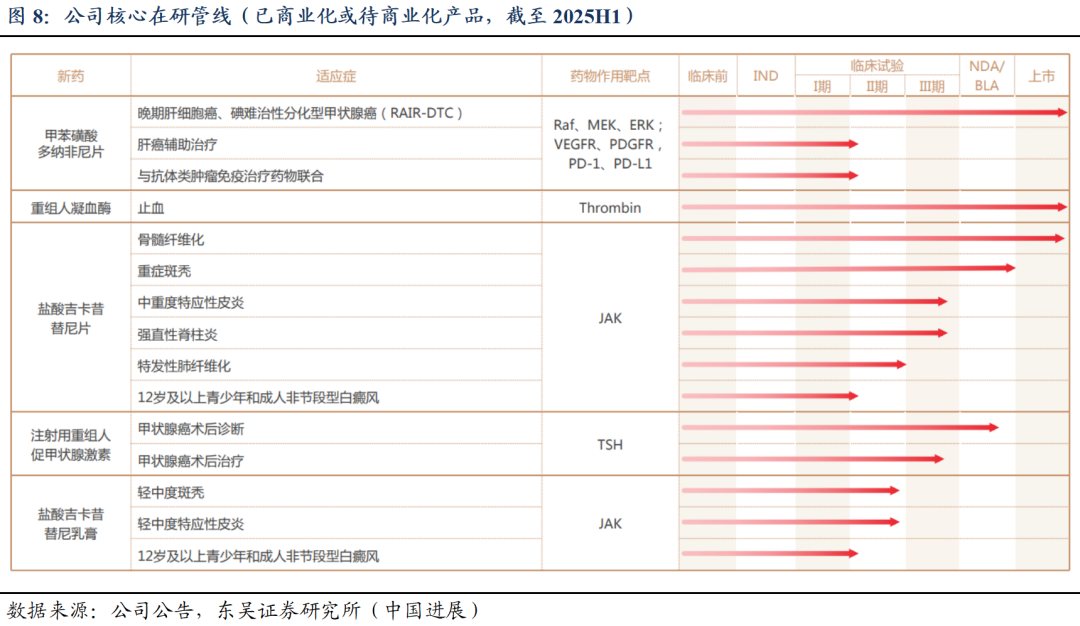
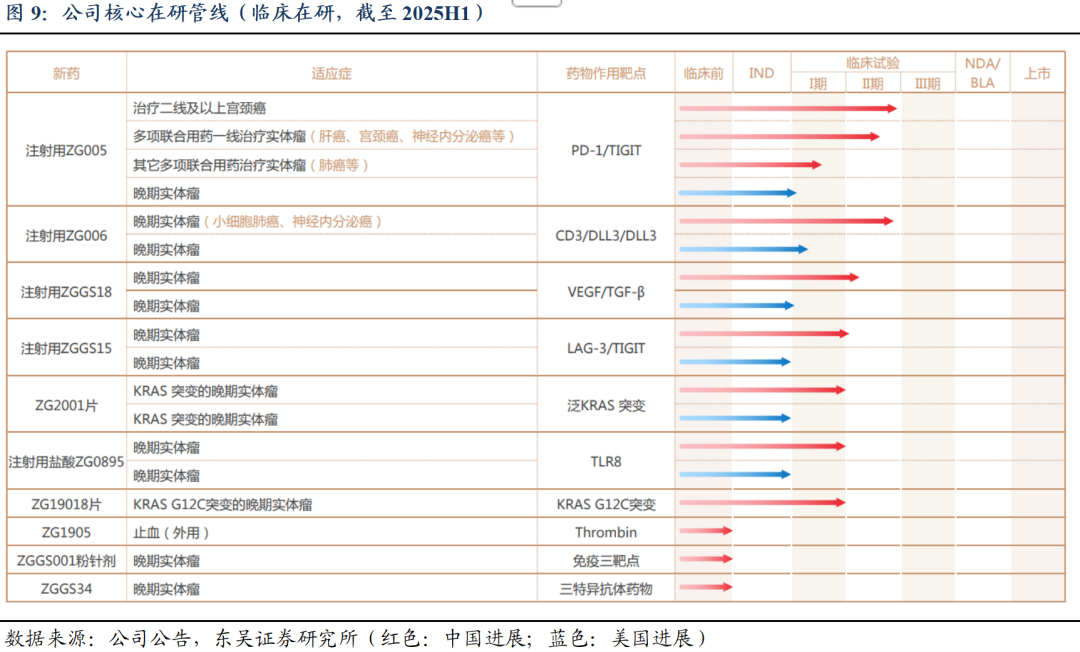
2. ZG006:结构新颖的DLL3三抗,有望打造SCLC基石疗法
2.1.DLL3为SCLC最具潜力的治疗靶点之一
小细胞肺癌(SCLC)属于神经内分泌瘤,占所有新发肺癌患者的13%~15%,具有生长快、侵袭性高、转移早、预后差等特点,约80%~85%的患者首次确诊时处于广泛期阶段(ES-SCLC)。据CACA前沿, 2022年我国肺癌新发病例约106.1万例(对应SCLC患者数约15万例)。SCLC现有效治疗方案疗效有限,晚期患者5年生存率只有7%,具有很大的未满足治疗需求。
DLL3为目前最具潜力治疗靶点之一。德尔塔样配体3(DLL3)作为Notch信号通路的唯一抑制性配体,通过与Notch受体相互作用抑制Notch信号通路的激活,参与肿瘤增殖、迁移和侵袭等过程。DLL3在正常细胞中极低表达,而在小细胞肺癌、神经内分泌癌等肿瘤细胞中高表达,因此DLL3为当前最具吸引力的选择性治疗靶点之一。
DLL3作为创新型抗肿瘤药物已成专家共识。《小细胞肺癌免疫治疗专家共识(2025版)》指出,对于复发SCLC的新免疫治疗方案中,推荐符合标准的SCLC患者加入靶向DLL3的BiTEs、双抗、BTLA抑制剂等新药的临床试验;《2025 CSCO小细胞肺癌诊疗指南》提及靶向DLL3/CD3双特异T细胞连接器抗体、抗体偶联药物等药物研发为SCLC患者带来新的希望,未来有望实现更大的突破。
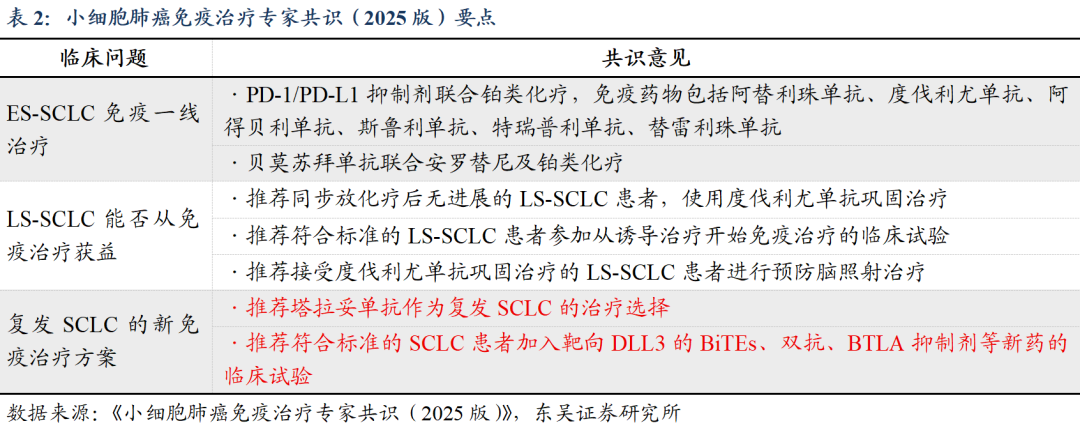

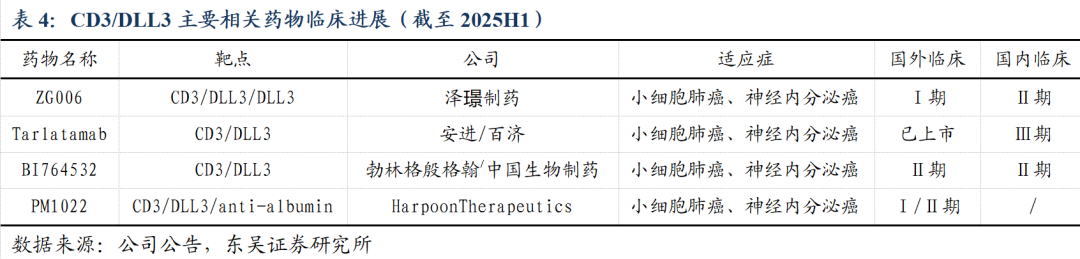
DLL3全球进展(统计截至2025H1):
安进的CD3/DLL3双抗塔拉妥单抗(Tarlatamab)针对末线SCLC患者临床试验的疗效突出,使得后线患者mOS由8个月提高至14.3个月,因而于2024年5月获得美国FDA加速批准上市;紧随其后为勃林格殷格翰/中国生物制药的CD3/DLL3双抗(国内外均为Ⅱ期)、公司的CD3/DLL3/DLL3三抗(国内Ⅱ期、国外Ⅰ期)。除了双抗/三抗之外,再鼎医药以及百利天恒的 DLL3 ADC、传奇生物的DLL3 CAR-T 等均已步入临床阶段。
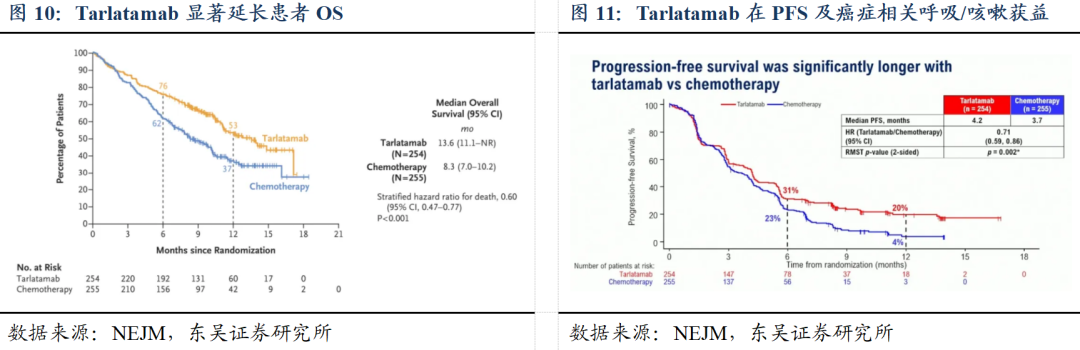
他山之石:塔拉妥单抗于2025ASCO大会上口头报告其Ⅲ期临床结果,显示OS获益。针对DeLLphi-304临床试验(多国、III期、开放标签的临床试验,比较Tarlatamab与化疗在小细胞肺癌患者中作为二线治疗的效果),共有509名患者被随机分配至Tarlatamab组(254人)或化疗组(255人)。Tarlatamab显著延长了患者的总生存期(中位OS为13.6个月,化疗组为8.3个月;分层死亡风险比为0.60;95%置信区间为0.47至0.77;P 0.001)。与化疗相比,Tarlatamab在无进展生存期及癌症相关的呼吸困难和咳嗽方面也表现出显著获益。3级及以上不良事件的发生率在Tarlatamab组低于化疗组(54%vs.80%),因不良事件导致停药的发生率也较低(5%vs.12%)。
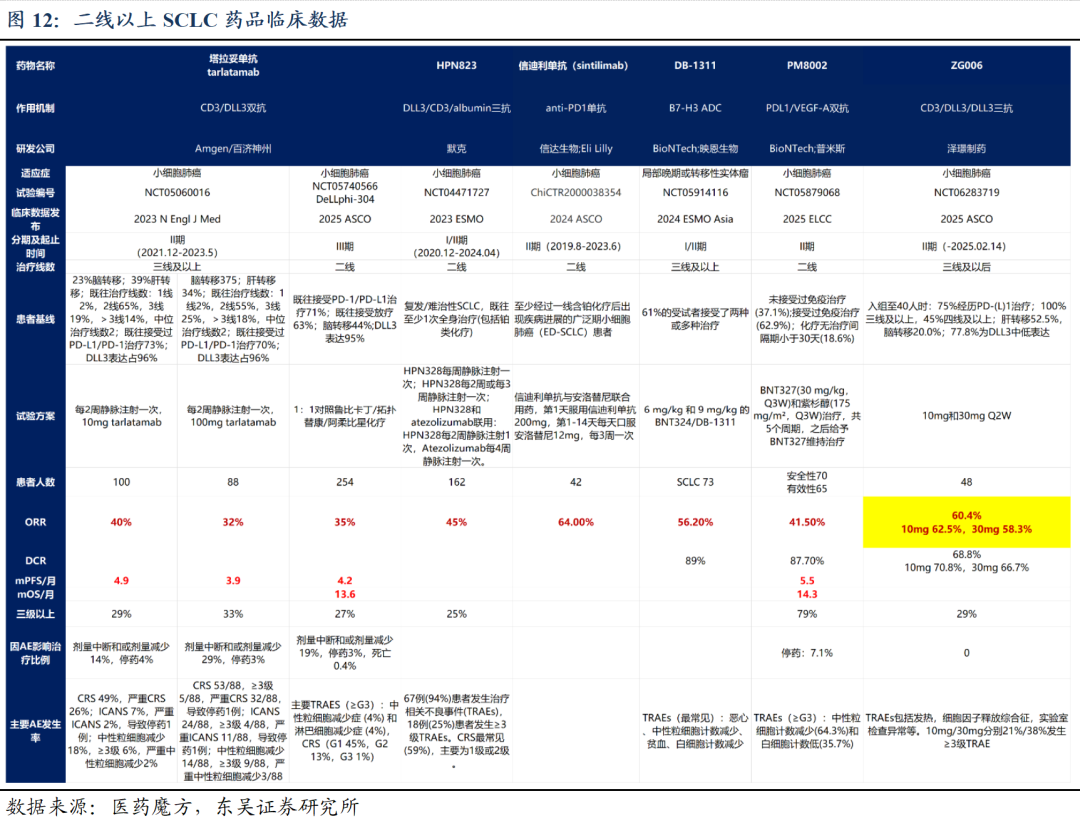
2.2 ZG006:疗效初步确认,有望成为SCLC领域最佳治疗药物
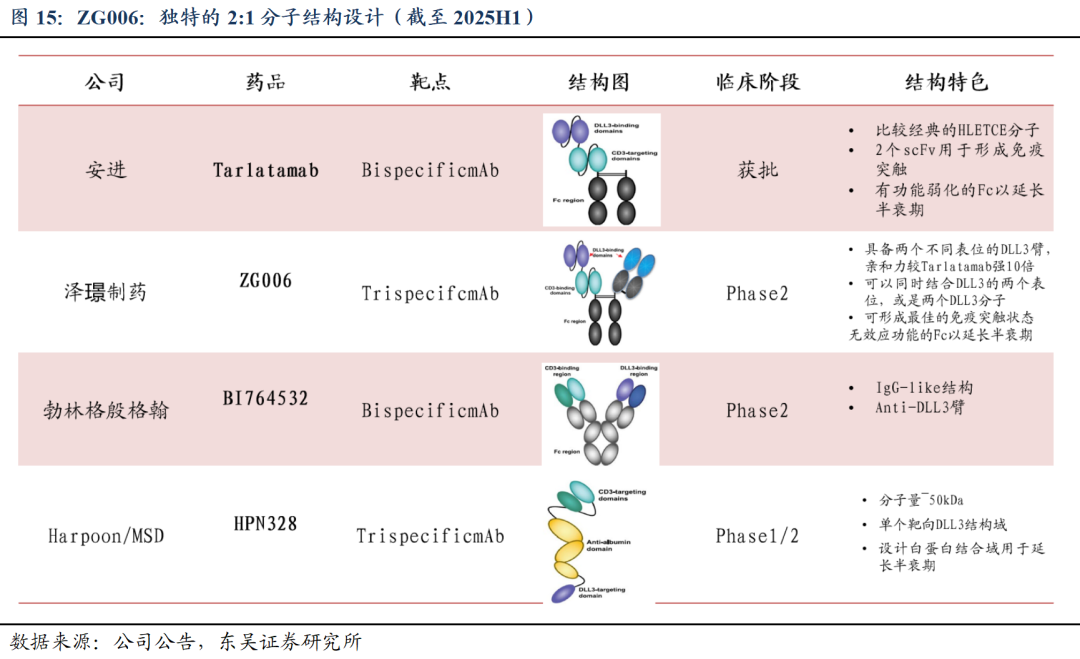
ZG006为公司独特设计的新一代TriTE分子,结构全球唯一、进展同类前三。独特的2:1设计能够结合两个不同表位的DLL3,表现出对DLL3很强的结合亲和力,具备优异的体外T细胞激活功能和杀伤能力,且抗体无功能Fc端,能够延长药物半衰期。
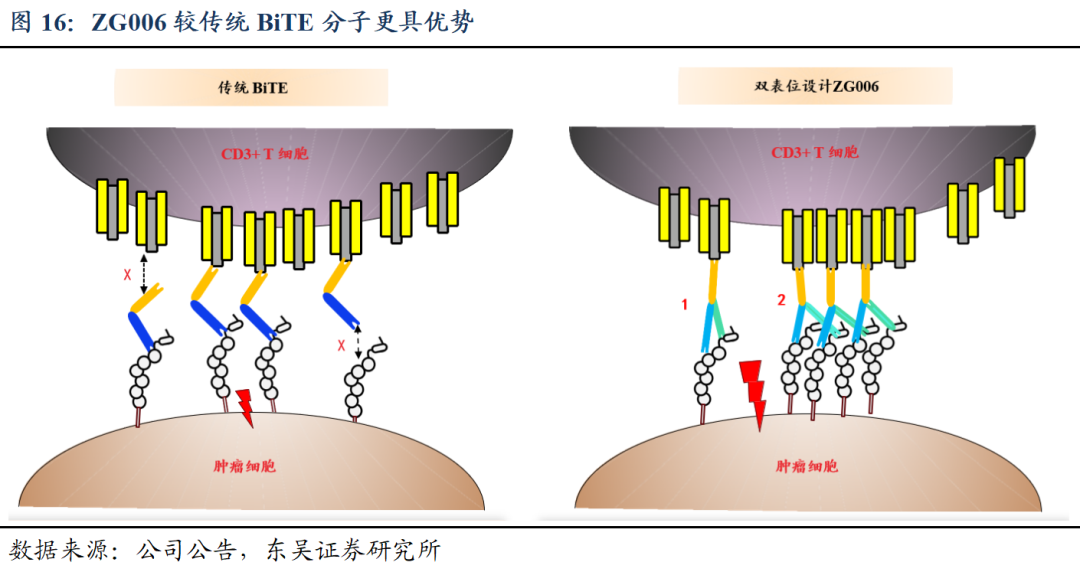
ZG006较传统BiTE分子更具优势。传统BiTE有时较难形成稳定的免疫突触,尤其是在DLL3低表达环境中,因此具备局限性;而ZG006可以结合在一个DLL3分子的两个表位上(紧密结合)或是结合在不同DLL3分子的两个表位上(聚集效应),起到协同作用从而形成稳定的免疫突触,并且增加结合强度。
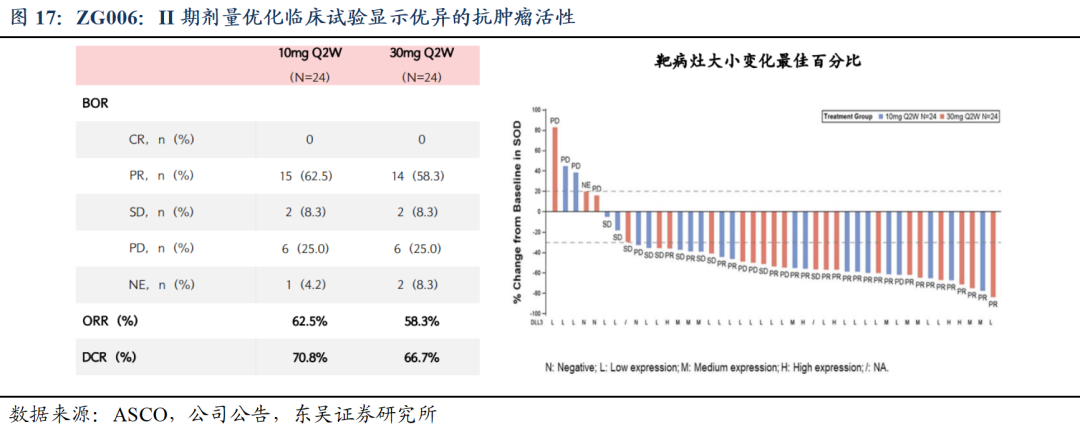
ASCO口头汇报,疗效数据优异,打造小细胞肺癌基石疗法。截至2025ASCO,ZG006针对小细胞肺癌II期临床进入尾声,III期注册临床方案沟通中;针对三线以上小细胞肺癌、ADC耐药患者以及一线联合疗法都将开展注册临床,全面布局小细胞肺癌赛道,打造基石疗法。
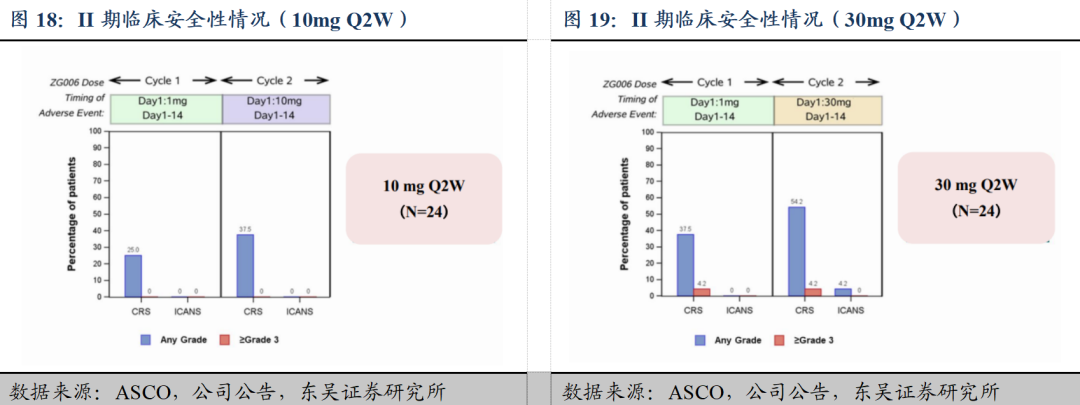
1)ZG006单药治疗在难治性晚期小细胞肺癌患者中的II期剂量优化临床研究(ZG006-002)入选本次年会肺癌专场口头报告: 10 mg Q2W和30 mg Q2W剂量在三线及以上小细胞肺癌患者中均展现出显著的抗肿瘤活性及良好的安全性。
截至2025年2月14日数据分析集,共48例三线及以上SCLC患者按1:1随机接受ZG006 10 mg Q2W或30 mg Q2W治疗并完成至少一次疗效评估,首次给药均为1 mg滴定剂量。两组患者的基线特征总体均衡。①有效性方面,基于IRC评估,10 mg和30 mg组的ORR分别为62.5%和58.3%,DCR分别为70.8%和66.7%; mPFS和mDoR尚未成熟。此外,在DLL3低表达患者或基线脑转移患者中都展现出良好的抗肿瘤活性。②安全性方面,两组的整体耐受性和安全性均良好,未发生任何因TEAE导致的永久停药。常见TRAE为发热、CRS及实验室检查异常,绝大多数TRAE为1-2级。10 mg和30 mg组分别有5例和9例患者发生≥3级TRAE。此外,绝大多数CRS为1-2级,主要发生于前两个治疗周期,对症治疗后大多可迅速恢复。
2)ZG006在晚期小细胞肺癌或神经内分泌癌患者中的耐受性、安全性、有效性和药代动力学的剂量递增和扩展的 I/II 期临床研究(ZG006-001)数据及最新进展:在单药治疗中展现出良好的安全性、耐受性及优异的抗肿瘤活性,为其进一步临床研究奠定了坚实基础。
截至2025年2月14日,在ZG006-001剂量递增及扩展研究中,共47例患者(43例小细胞肺癌患者、4例神经内分泌癌患者)接受了从0.1 mg至100 mg不等剂量的ZG006 Q2W治疗,其中10 mg及以上剂量的首次给药为1 mg滴定剂量。所有患者均完成了至少一次疗效评估,包括≤3 mg 13例、10 mg 5例、30 mg 14例、60 mg 12例、100 mg 3例。①有效性方面,根据研究者评估,ZG006单药在多个剂量水平治疗晚期小细胞肺癌患者展现出良好的抗肿瘤活性,特别是10 mg、30 mg和60 mg Q2W剂量,确认的ORR分别为75.0%、53.8%和58.3%,DCR分别为75.0%、76.9%和83.3%。评估截止时间时,PFS和DoR还未成熟。②安全性方面,47例患者均发生了TRAE,大多数为1-2级。常见TRAE包括CRS、发热、实验室检查异常等,经对症治疗后多可恢复或缓解。

3)ZG006在晚期神经内分泌癌患者中的Ⅱ期剂量扩展临床研究(ZG006-003)数据及最新进展:在二线及以上神经内分泌癌患者中展现出显著的抗肿瘤活性及良好的安全性,支持其在该适应症中开展进一步的临床研究。
截至2025年3月21日,共46例二线及以上神经内分泌癌患者接受ZG006治疗,按1:1随机接受10 mg Q2W或30 mg Q2W治疗,首次给药均为1 mg滴定剂量。①有效性方面,10 mg Q2W和30 mg Q2W组分别有8例和9例疗效可评估受试者,根据研究者评估,未确认的ORR分别为12.5%和55.6%,DCR分别为37.5%和77.8%;mPFS和mDoR尚未成熟。②安全性方面,87.0%(40/46)例受试者发生了TRAE,绝大多数为1或2级。
3. ZG005:新型PD-1/TIGIT双抗,泛瘤种潜力有望成为重磅炸弹
3.1. PD-1/TIGIT双抗赛道进展顺利,阿斯利康Rilve已进入多项Ⅲ期临床
TIGIT是一种重要的免疫检查点分子,具有一个免疫球蛋白样域和一个免疫受体酪氨酸抑制基序(ITIM),属于免疫球蛋白超家族。TIGIT在NK细胞、T细胞和树突状细胞(DCs)上表达,TIGIT的主要功能是通过与其配体CD155和CD112的相互作用来传递抑制信号,抑制T细胞和NK细胞的活化和效应功能,减少炎症反应和免疫应答,同时帮助肿瘤细胞逃避免疫监视。通过阻断TIGIT与其配体的结合,可以恢复或增强T细胞和NK细胞的抗肿瘤活性,从而提高免疫治疗的效果。因此,TIGIT成为了癌症免疫治疗的一个新兴靶点。
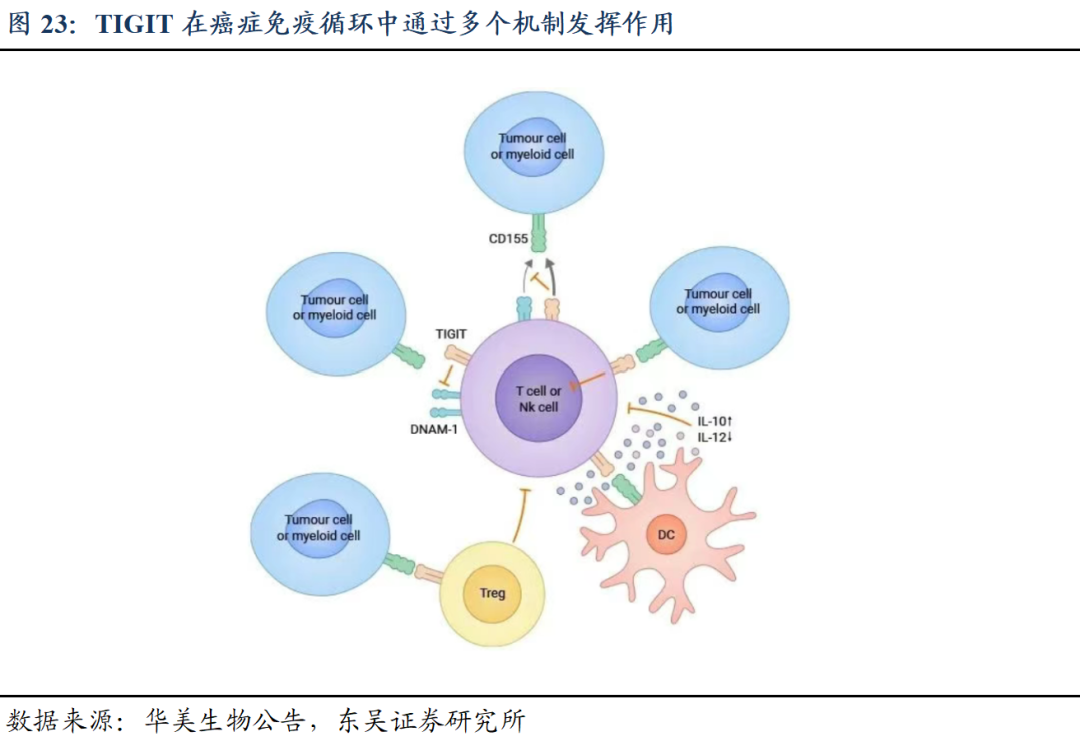
TIGIT单一阻断效果有限,多与PD-1/PD-L1联用。由于TIGIT单药治疗有局限性,且TIGIT受体在T细胞及NK细胞表达,同时靶向PD-1及TIGIT受体,具有更好抗肿瘤协同作用,因此目前全球涉及TIGIT的临床研究中,多为与PD-1/PD-L1联用,包括1)同时给药抗TIGIT和抗PD-1/PD-L1药物(例如tiragolumab+atezolizumab)。2)联合使用抗TIGIT和抗PD-1/PD-L1药物(如MK-7684A)。3)结合TIGIT和PD-1/PD-L1的双特异性抗体(如IBI321)。

PD-1/TIGIT双抗赛道进展顺利,阿斯利康Rilve已进入多项Ⅲ期临床。阿斯利康的Rilvegostomig是一款靶向PD-1+TIGIT的双特异性抗体,截至2025H1处于Ⅲ期临床阶段,在PD-1/TIGIT双抗赛道排名第一,并布局多项适应症,已启动9项III期临床,涵盖肺癌、胆道癌、肝癌、胃癌等。全球同靶点在研双抗为7款,国内看,公司的ZG005已进入Ⅱ期临床。

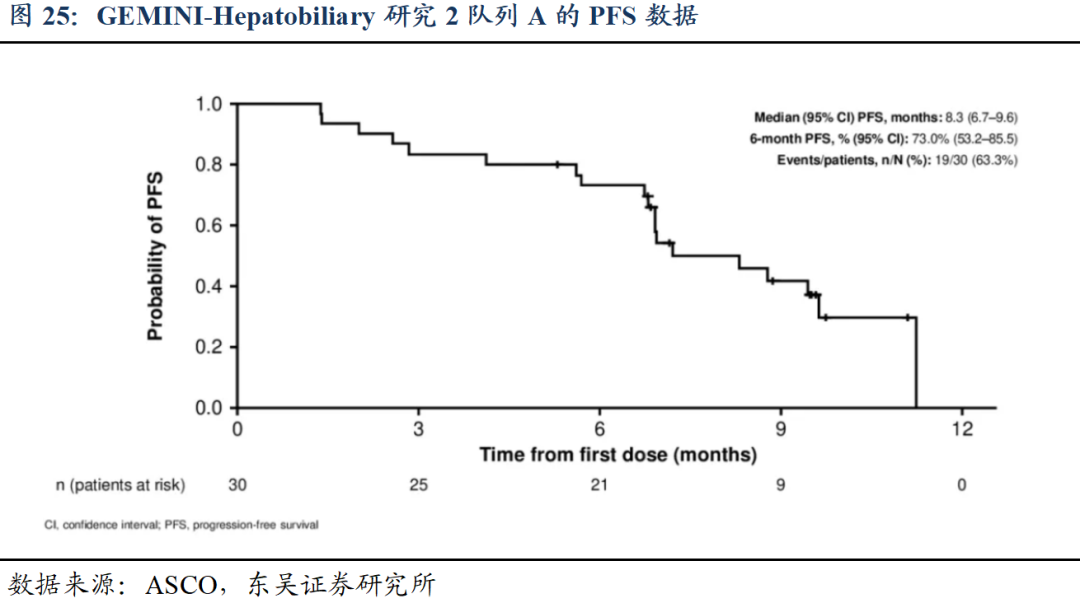
他山之石:2025年ASCO会议报告了GEMINI-Hepatobiliary研究2队列A(rilvegostomig联合化疗治疗BTC)的数据:rilvegostomig联合化疗方案在晚期BTC中展现出有前景的疗效和可控的安全性,并能持续实现靶点占据,其独特的PD-1/TIGIT双靶点结合机制为患者提供了新的治疗选择。
1)截至2024年11月4日,共30例患者接受了治疗,中位年龄为60岁,70.0%为亚洲人群,83.3%为转移性疾病。Rilvegostomig中位治疗周期数为10.5次,36.7%的患者仍在接受rilvegostomig治疗。吉西他滨/顺铂的治疗中位周期数均为8次,30%的患者仍在接受化疗。中位随访6.9个月时,6个月PFS率达73.0%(95%CI:53.2-85.5),中位PFS为8.3个月(95%CI:6.7-9.6)。OS数据尚未成熟(中位随访9.8个月时),9个月OS率为85.0%(95%CI:64.6-94.2)。
2)在29例可评估患者中,确认的ORR为31.0%(95%CI:15.3-50.8)。PD-L1肿瘤区域阳性评分(TAP)≥1%的患者(n=18)ORR为60%,PD-L1 ATP<1%的患者(n=9)ORR为30%。
3)安全性方面,不良反应谱主要由化疗导致,最常见的治疗相关不良事件(TRAE)包括贫血(53.3%)、中性粒细胞减少(50.0%)和血小板减少(43.3%)。Rilvegostomig相关AE主要为谷丙转氨酶(ALT)升高(20.0%)和皮疹(13.3%)。≥3级免疫相关不良事件发生率为13.3%,贫血、血碱性磷酸酶升高、脂肪酶升高和肺炎各1例。
3.2. ZG005:早期数据优异,适应症拓展潜力较大
ZG005为新型抗PD-1/TIGIT双特异性抗体,当前处于Ⅱ期临床,中美双报。该抗体设计1)能够同时靶向PD-1及TIGIT,具有更好协同作用,激活T细胞及NK细胞杀伤效果,临床前数据显示疗效优于单药及单药联合。2)IgG4结构减少ADCC效应,避免T细胞及NK细胞消耗,强化T细胞及NK细胞的杀伤效果。3)促进TIGIT的共刺激受体CD266表达,增强杀伤效果;减轻Treg细胞介导的免疫抑制。

ASCO披露详细II期数据,初步疗效已验证,早期数据优异且适应症拓展潜力较大,有望成为下一个重磅品种。ZG005针对宫颈癌二三线已完成所有患者入组,III期双抗头对头注册临床方案和CDE沟通中;一线肝癌II期同步推进中,初步疗效信号较好;联合JAK开发PD1/PDL1经治的非小细胞肺癌获批临床,适应症开发潜力较大。
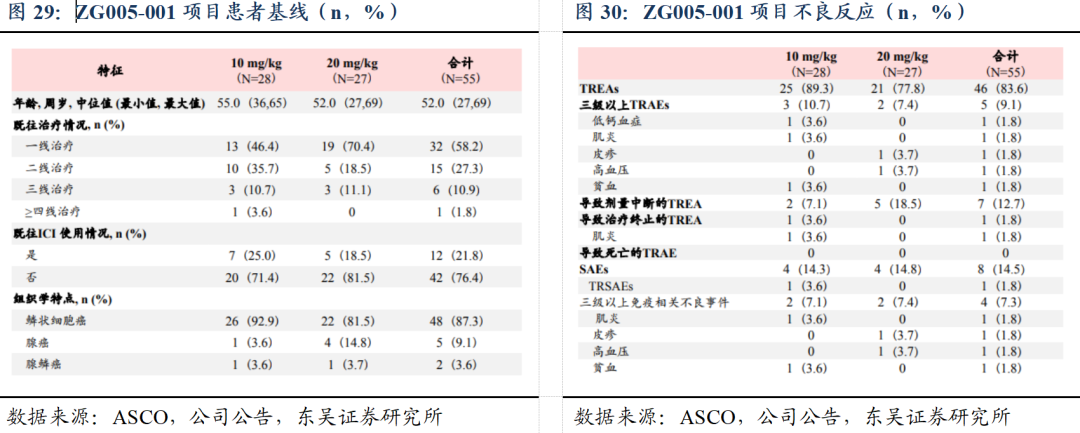
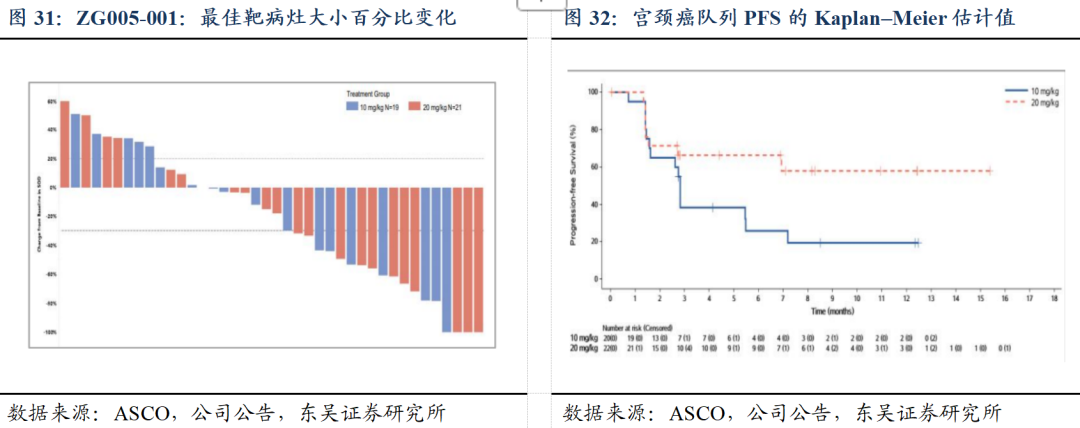
1)ZG005在晚期实体瘤患者中的剂量递增、耐受性、安全性、药代动力学和多队列扩展的 I/II 期临床研究(ZG005-001)数据及最新进展:20 mg/kg Q3W给药剂量下,在二线及以上宫颈癌患者中,展现出优异的抗肿瘤活性和良好的安全性。
截至2024年12月5日,ZG005-001项目I期剂量递增阶段已完成;II期剂量扩展阶段中,共55例二线及以上宫颈癌患者接受(ZG005)治疗,按1:1随机接受10 mg/kg Q3W或20 mg/kg Q3W治疗。①有效性方面,既往未接受过免疫检查点抑制剂治疗的二线及以上宫颈癌患者中,20 mg/kg组(N=22)基于IRC评估的确认ORR为40.9%,DCR为68.2%。基于研究者评估,20 mg/kg组mPFS已超过11个月。②安全性方面,两组整体耐受性和安全性均良好,83.6%的患者发生TRAE,绝大多数为1-2级,仅9.1%为≥3级,未发生因TEAE导致的死亡。
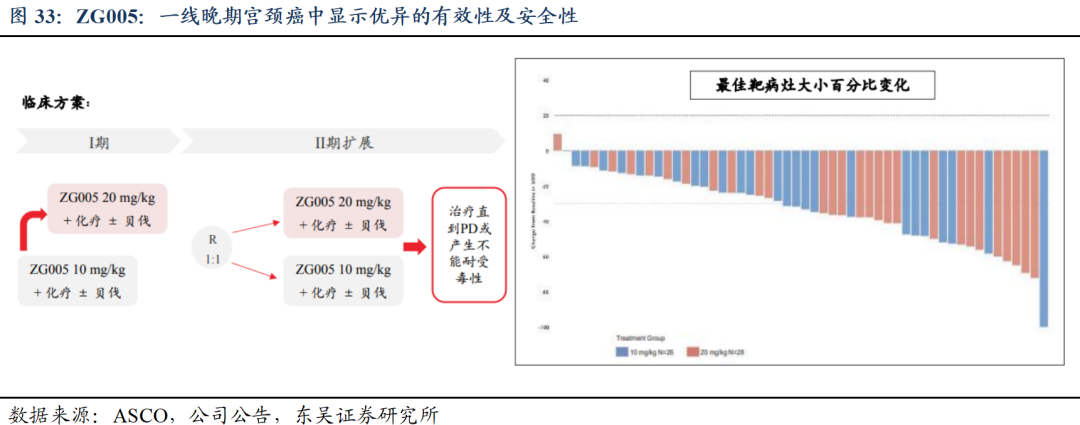
2)ZG005联合紫杉醇及铂类±贝伐珠单抗方案在晚期宫颈癌患者中的安全性、药代动力学特征及初步疗效的 I/II 期临床研究(ZG005-003)数据及最新进展:对一线宫颈癌展现出良好的疗效,特别是20 mg/kg剂量组有效性突出;同时,联合治疗在两个剂量水平上均具有良好的安全性和耐受性,支持其进一步研究和临床应用。
截至2025年4月23日,共入组60例一线宫颈癌患者,其中I期剂量递增阶段已完成,共纳入13例患者;II期剂量扩展阶段正在进行中,已有47例患者接受了ZG005治疗。按1:1随机接受了10 mg/kg Q3W或20 mg/kg Q3W联合紫杉醇及铂类±贝伐珠单抗治疗。随机分层因素为既往是否接受过贝伐珠单抗治疗,60.0%的患者在本试验中联合使用了贝伐珠单抗。①有效性方面,基于研究者评估,20 mg/kg组(N=28)的未确认ORR为82.1%,DCR为96.4%;10 mg/kg组(N=26)的未确认ORR为65.4%,DCR为96.2%。两组mPFS和mDoR均尚未成熟。②安全性方面,两组的耐受性和安全性均良好,88.3%发生了TRAE,绝大多数为1-2级,3-4级TRAE发生率为45.0%,两组均未发生任何与ZG005相关的永久停药或死亡。
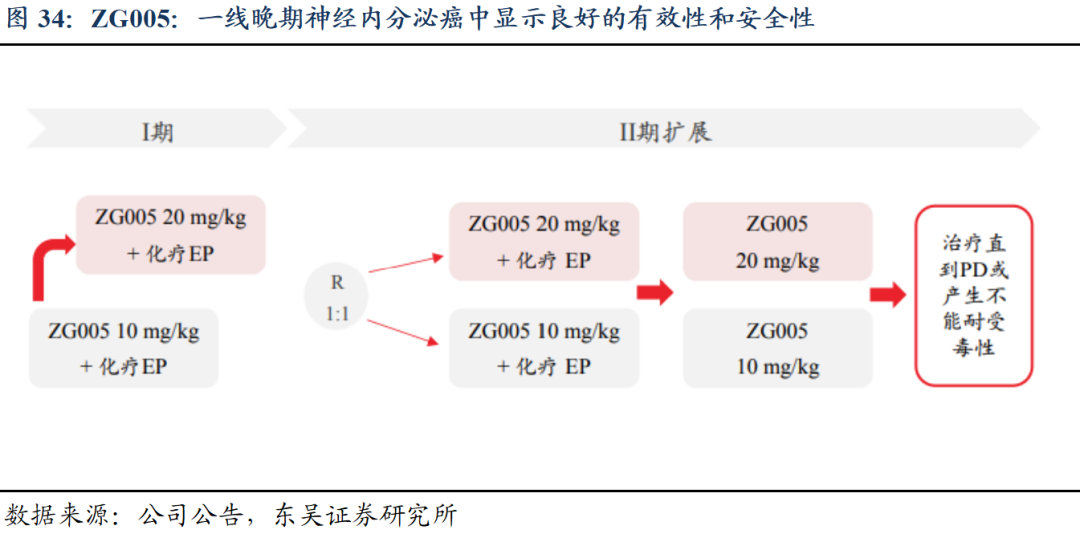
3)ZG005联合依托泊苷及顺铂在晚期神经内分泌癌患者中的安全性、药代动力学特征及初步疗效的I/II期临床研究(ZG005-004)数据及最新进展:在晚期神经内分泌癌的患者中,ZG005联合化疗安全性耐受性良好,显示了良好的疗效,支持其开展进一步的临床研究。
截至2025年1月10日,共纳入21例患者。所有患者Ki-67指数均≥55%,66.7%的患者存在肝转移。最常见的原发肿瘤部位为胃肠道,占38.1%。中位治疗周期为4个周期(2-4个周期)。①有效性方面,在12例可评估疗效的患者中,6例部分缓解(PR)(其中4例为确认的PR),其中2例接受ZG005 10 mg/kg + EP方案,4例接受ZG005 20 mg/kg + EP方案;另有5例患者病情稳定(SD)。ORR为50%,DCR为91.7%。②安全性方面,在Part 1剂量递增阶段未观察到剂量限制性毒性(DLT)。最常见的TRAE包括贫血、白细胞计数减少、中性粒细胞计数减少和丙氨酸氨基转移酶升高。3例患者发生≥3级的TRAE。4例患者发生严重不良事件(SAEs),其中仅免疫介导性肠炎 1例与ZG005相关。
4.商业化产品梯次落地,创新硕果空间广阔
4.1.多纳非尼:首款国产肝癌靶向药,目前已有2个适应症获批上市
甲苯磺酸多纳非尼片是公司自主研发的口服小分子多靶点多激酶抑制剂,于2021年6月获批上市,是公司的第一款商业化产品。该药物既可通过抑制血管内皮生长因子受体(VEGFR)和血小板源性生长因子受体(PDGFR)等多种酪氨酸激酶受体的活性,阻断肿瘤血管生成,又可通过阻断丝氨酸-苏氨酸激酶(RAS/RAF/MEK/ERK)信号传导通路直接抑制肿瘤细胞增殖,从而发挥双重抑制、多靶点阻断的抗肿瘤作用。2022年8月,多纳非尼治疗局部晚期/转移性分化型碘难治性分化型甲状腺癌(RAIR-DTC)获批上市。一线晚期肝癌适应症通过2023年国家医保谈判,继续纳入国家医保目录,并新增用于甲状腺癌患者的适应症范围。
晚期肝癌患病人群广、市场空间大。根据弗若斯特沙利文,2020年我国肝癌新发病例数达42.1万例,预计2030年新发病数达52.7万例,且当期我国肝癌药物市场规模有望达452.1亿元。

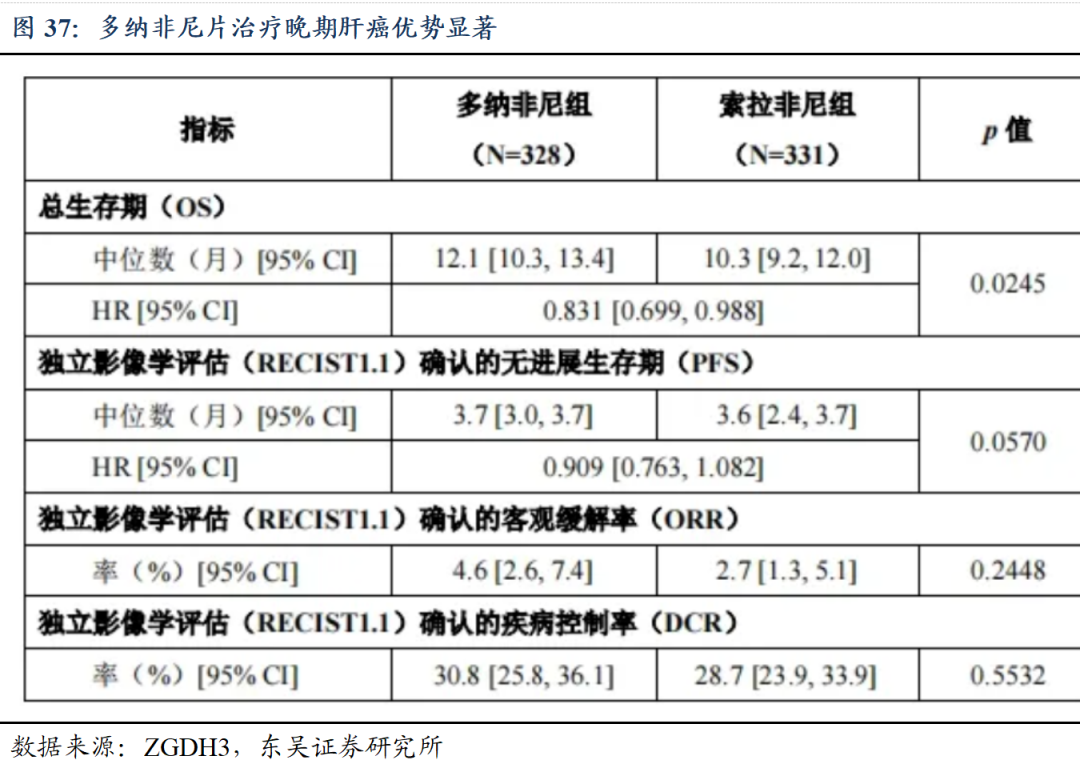
多纳非尼片治疗晚期肝癌优势显著。作为唯一一个单药与索拉非尼头对头临床试验取得优效的晚期肝癌一线治疗新药,与索拉非尼相比,多纳非尼显著延长晚期HCC患者的总生存期;安全性显著优于索拉非尼。
我国RAIR-DTC治疗存在未被满足的临床需求。局部晚期/转移性分化型碘难治性分化型甲状腺癌(RAIR-DTC)患者具有死亡高风险性,10年生存率为10%,平均预期生存时间仅为3-5年。
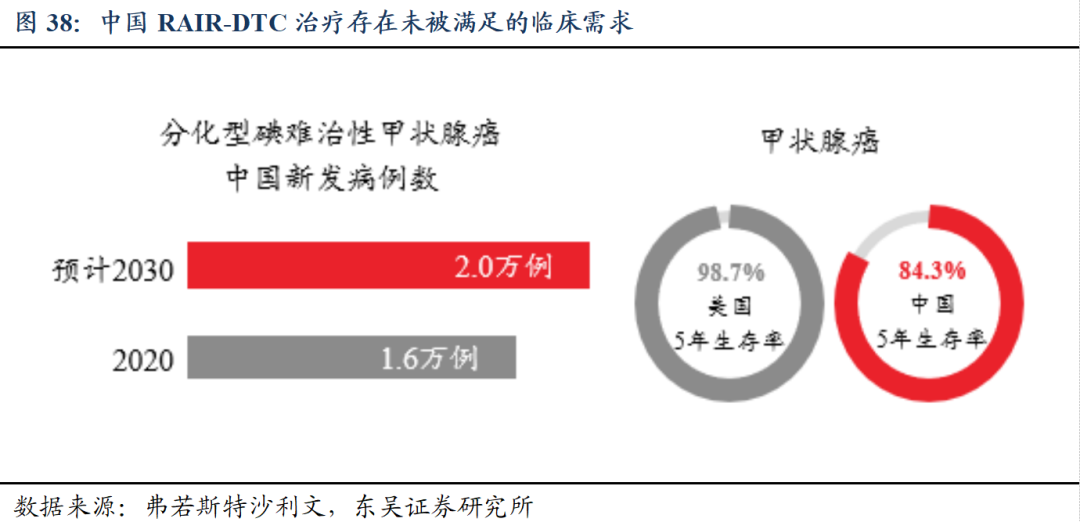
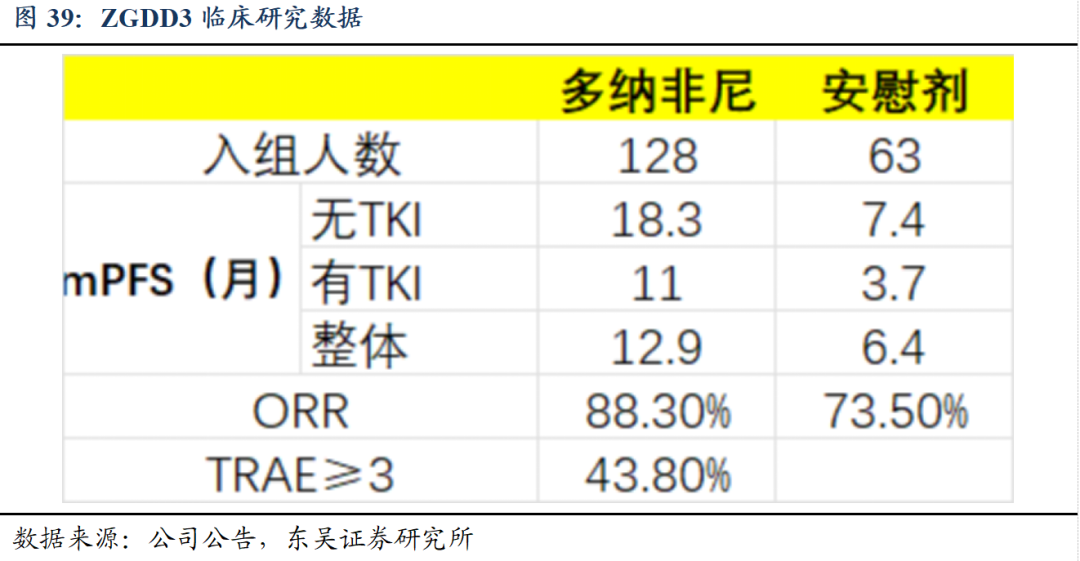
多纳非尼片治疗RAIR-DTC疗效显著。III期临床试验主要有效性结果显示:多纳非尼组较安慰剂组可以显著降低疾病进展的风险,显著延长无疾病进展生存期(延长一倍以上),同时获得更佳的客观缓解率,具备疗效和安全性优势。
公司积极推动产品商业化,多纳非尼纳入医保和指南,持续扩大市场份额。截至2024年底,多纳非尼两款适应症均纳入国家医保药品目录,实现进院1100+家,覆盖医院2000+家,覆盖药房近1000+家,纳入国内权威26个指南/共识,全国省份100%覆盖。

4.2.重组人凝血酶:首个重组人凝血酶产品,满足迫切临床需求
我国外用手术止血药物市场规模大,尚无同类产品上市。根据弗若斯特沙利文,预计我国到2030年外科手术约1.28亿台,对应外科手术出血局部药物市场为161.6亿元。
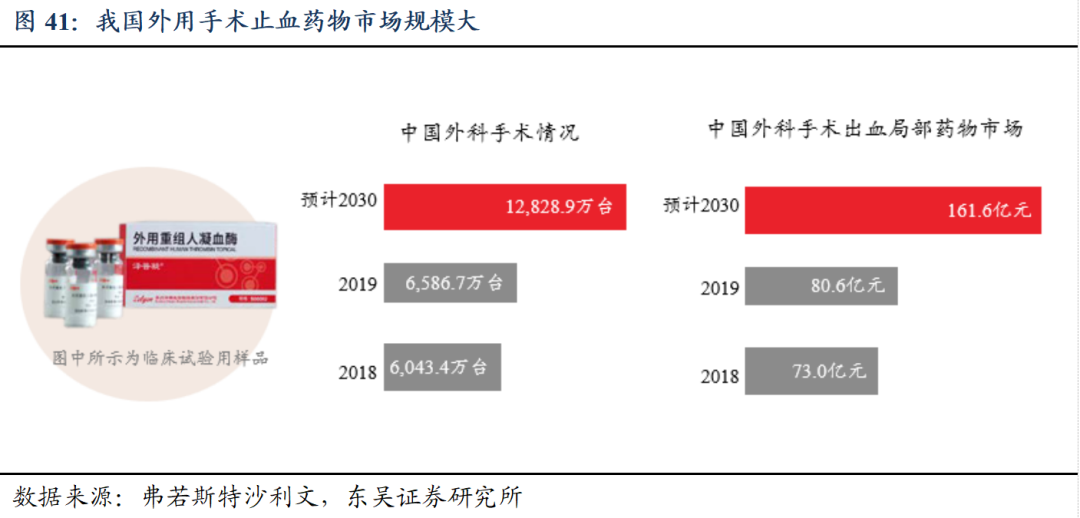
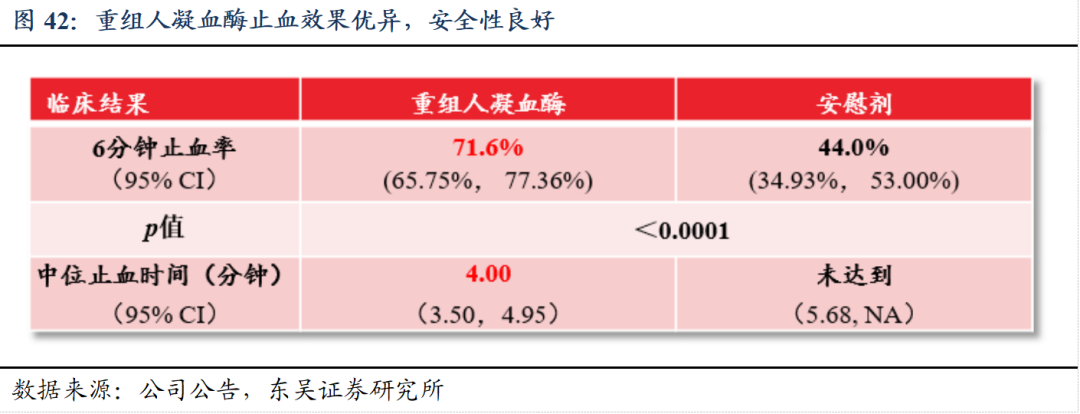
重组人凝血酶为国内首个获批的新一代局部止血药品,满足迫切的临床需求。中国现有的外科手术局部止血药物主要为人血来源/动物来源凝血酶、纤维蛋白原、蛇毒血凝酶等,动物源性及人源性的凝血酶均存在一定的安全性风险,动物源性的还可能存在较高免疫原性风险。而重组人凝血酶具有更高的生物安全性优势和更高的凝血活性,可大规模生产且成本可控。重组人凝血酶是中国首个获批的新一代局部止血药品,具有突出的临床止血效果及良好的安全性,并已成功纳入《国家基本医疗保险、工伤保险和生育保险药品目录(2024年)》。
公司重组人凝血酶与远大生命科学达成独家商业化合作。远大生命科学是一家深耕止血(远大辽宁)、疫苗(远大赛威信)、血制品(远大蜀阳)等领域的综合性医药企业集团,尤其是远大辽宁在止血、麻醉镇痛及创面管理等领域具备领先优势,以注射用矛头蝮蛇血凝酶为代表的止血产品连续多年销量处于国内领先地位。远大拥有外科领域成熟的商业化团队,目前积极开展市场教育及准入工作。本次合作首付款和商业化里程碑款总金额4亿元,后续达到协议约定的销售里程碑事件后,最高不超过9.15亿元人民币销售里程碑款。
4.3.吉卡昔替尼:MF适应症获批,自免商业化空间广阔
吉卡昔替尼片一类JAK激酶小分子抑制剂,为公司自主研发的靶向小分子I类新药。作为广谱JAK激酶小分子抑制剂,吉卡昔替尼对JAK1、JAK2、JAK3和TYK2具有显著抑制作用,通过阻断JAK-STAT信号通路,调节细胞因子信号传导,进而干预免疫炎症反应,在多种免疫炎症性疾病和血液疾病治疗中展现潜力。
骨髓纤维化(MF)存在未被满足的临床需求。根据弗若斯特沙利文,预计2030年国内骨髓纤维化患者年新发6.3万,存量患者数预计约30万人,靶向药市场的临床可惠及人口渗透率将达到30.7%。目前针对骨髓纤维化的治疗药物有限,以对症支持治疗为主。全球范围内仅有芦可替尼、菲达替尼和帕瑞替尼三款靶向药物获批用于骨髓纤维化,在国内仅有芦可替尼上市。2025年5月29日,公司盐酸吉卡昔替尼片MF适应症获批上市,有望迅速占据国内市场份额。
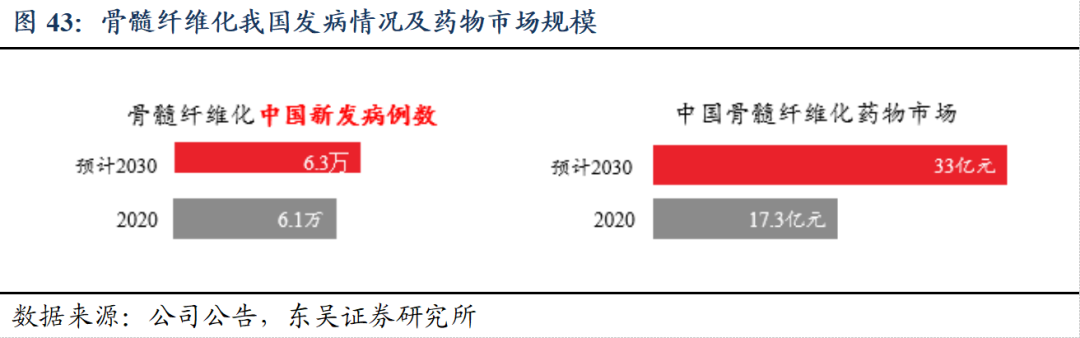
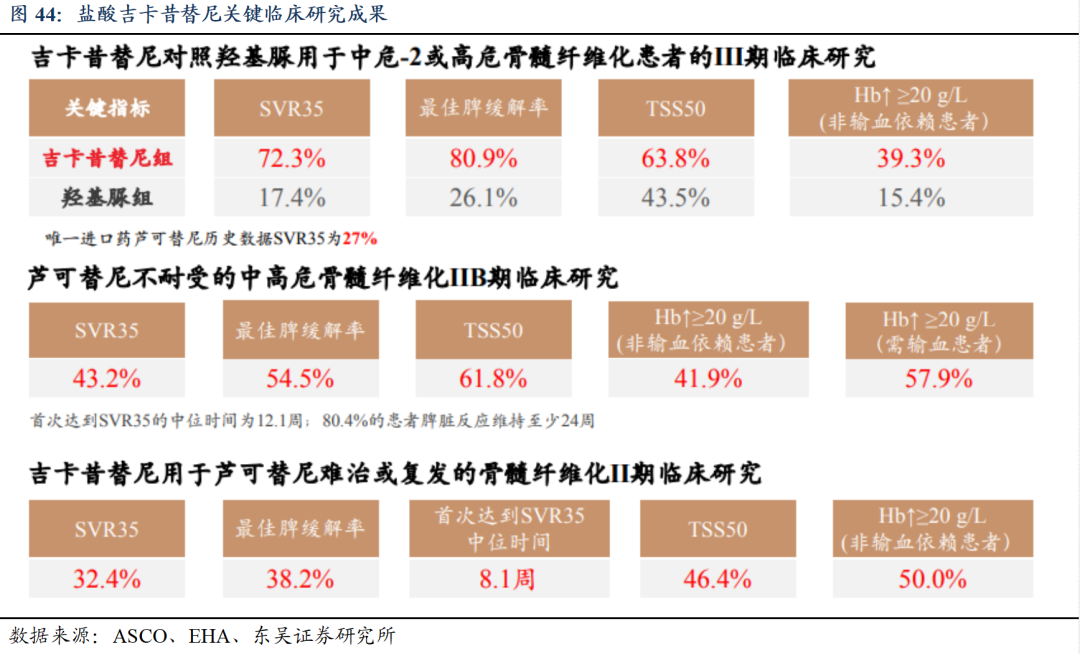
盐酸吉卡昔替尼片有望成为BIC药物。在《CSCO恶性血液病诊疗指南(2024年)》纳入了原发性骨髓纤维化(PMF)一线分层治疗的I级推荐,并维持二线及进展期治疗的推荐,尤其是在骨髓纤维化(MF)相关贫血患者的一线治疗中,被列为I级推荐的首选。公司正在美国开展复发难治性MF的I期临床试验,并获得FDA孤儿药资格。此外,盐酸吉卡昔替尼片受广泛认可,专利保护期长。此外,吉卡昔替尼关键指标改善程度显著高于其他已上市药物的临床试验数据,展现BIC潜力。
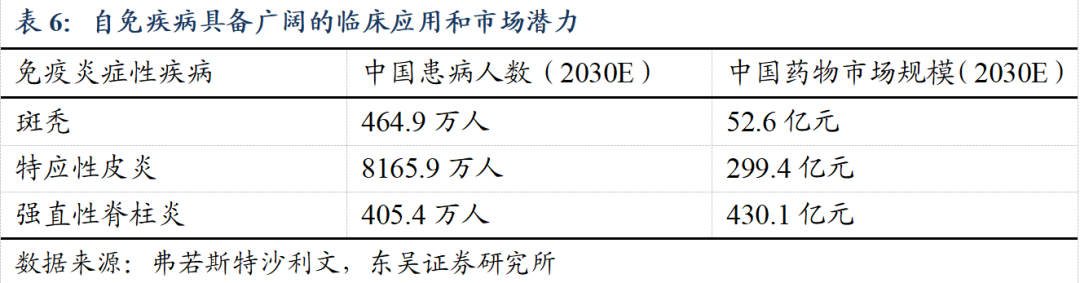
自免疾病市场规模庞大,公司吉卡昔替尼斑秃、特应性皮炎、强直性脊柱炎适应症均已进入Ⅲ期临床。据弗若斯特沙利文,我国2030年斑秃、特应性皮炎、强直性脊柱炎药物市场规模有望达52.6/299.4/430.1亿元。
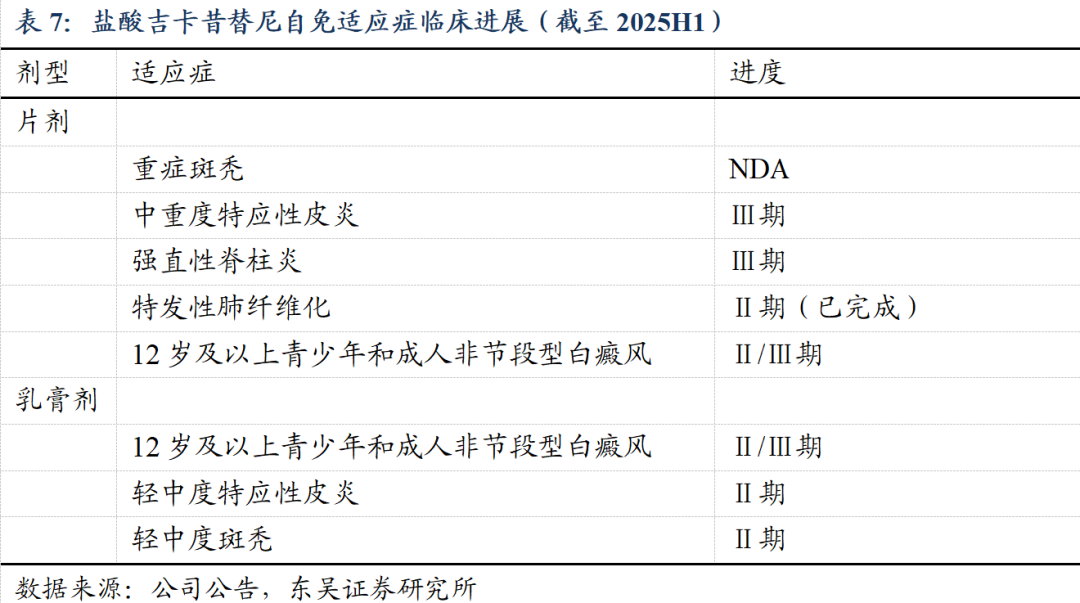
主要临床数据:
1)重症斑秃:
III期:主要疗效终点,即治疗24周脱发严重程度工具量表(SALT)评分≤20分的受试者百分率,吉卡昔替尼片两组均显著优于安慰剂组,达到统计显著性(p 0.0001)。
Ⅱ期:吉卡昔替尼片50mgBid、150mgQd和200mgQd三组的有效率(24周SALT评分较基线降低达50%及以上(SALT50)的受试者比率)分别为59.2%、63.3%和60.0%
安全性方面,吉卡昔替尼治疗重症斑秃患者的安全性与耐受性良好。
2)强直性脊柱炎:
II期:治疗16周时,100mgBid、75mgBid和安慰剂对照组ASAS20应答百分率分别为62.9%、59.4%和33.3%,两治疗组相对于安慰剂组p值均小于0.05,达到方案预设的统计学标准,结果稳健。
5.聚焦三大领域研发,在研产品潜力丰富
5.1 研发策略:构建肿瘤免疫和分子靶向联合治疗产品体系
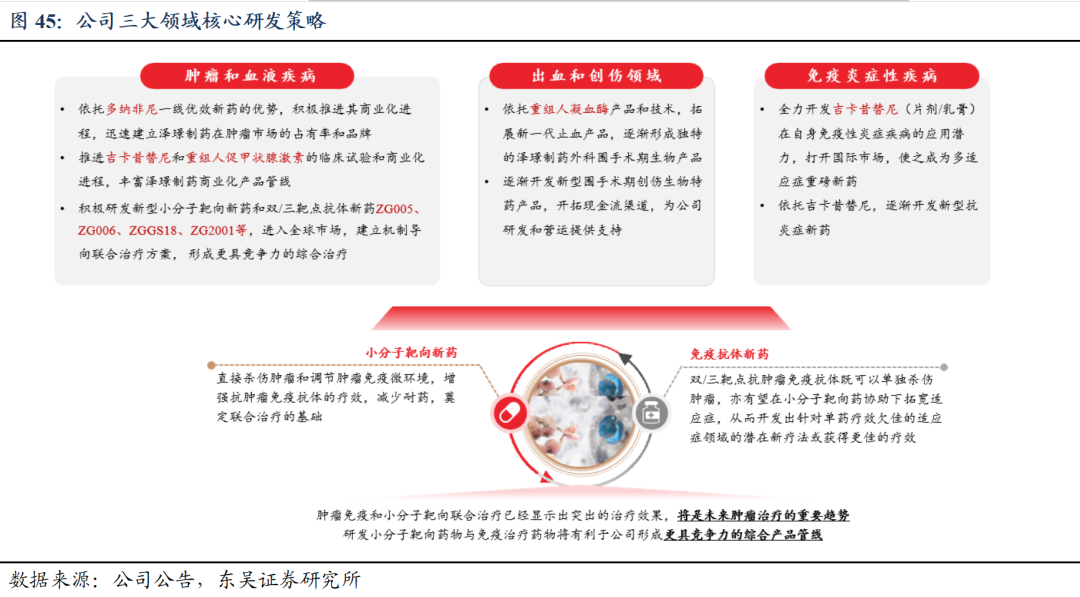
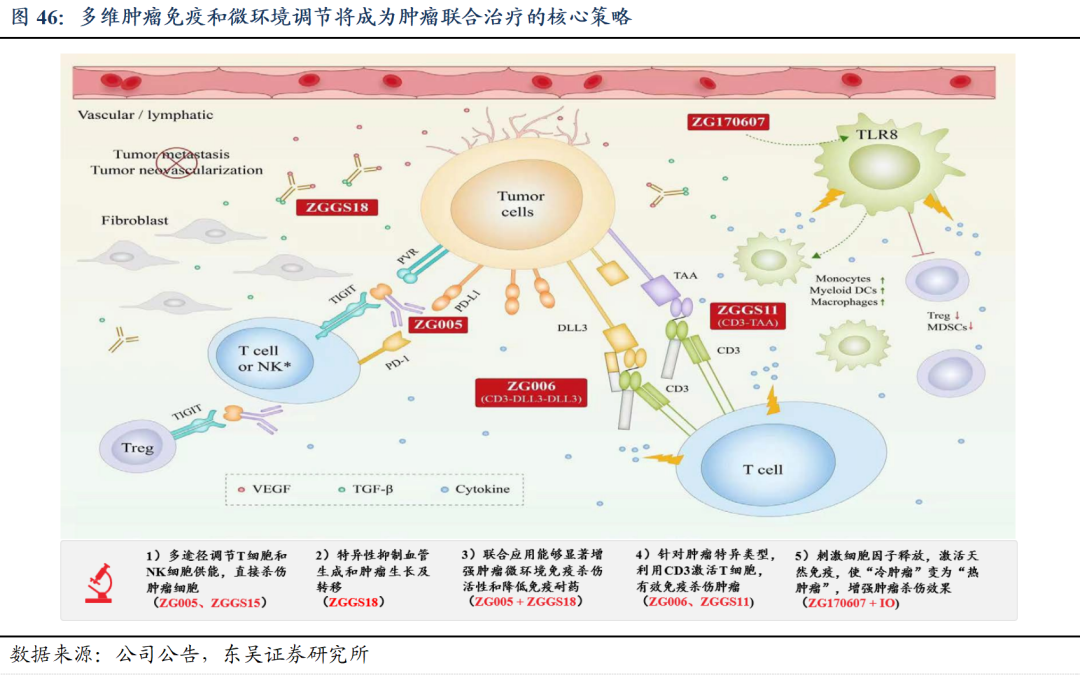
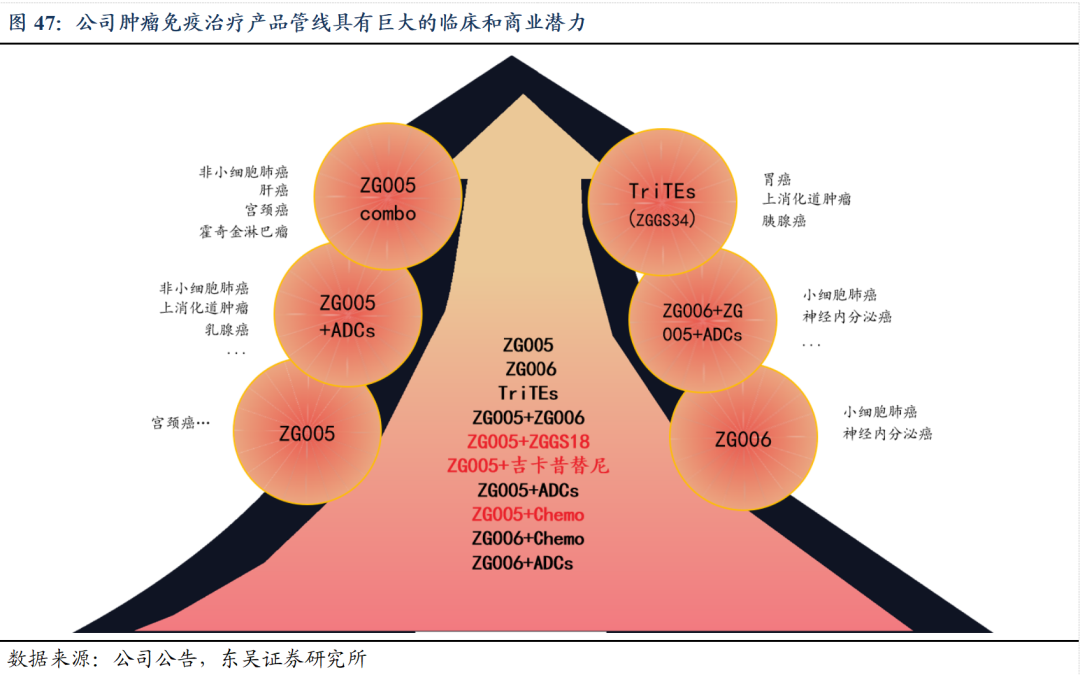
5.2.注射用人促甲状腺激素:填补甲状腺诊疗空白,市场空间广阔
2024年国家癌症中心发布的我国癌症统计报告显示:2022年我国甲状腺癌新发患者约46.61万人,是目前我国发病率排名第三的恶性肿瘤。注射用人促甲状腺激素(rhTSH)是中国首批取得用于甲状腺癌的术后诊断(已递交上市申请,已完成临床核查和二合一检查)和治疗(III期临床进行中)临床试验批件的新药,有利于进一步提高患者生存率和生存期。

分化型甲状腺癌确认后,对适合的患者将进行手术切除,随后实施I-131治疗。针对I-131治疗,需要甲状腺组织具备一定摄碘能力(即TSH≥30mIU/L),从而保障治疗效果,目前临床方案为注射rhTSH或者停服左甲状腺素钠片(L-T4),注射rhTSH方案已纳入最新指南推荐。在治疗后定期随访中,目前临床通过检测甲状腺球蛋白(Tg)水平,监测肿瘤的复发/转移,而Tg由TSH刺激后分泌,检测前需保障一定的TSH水平。
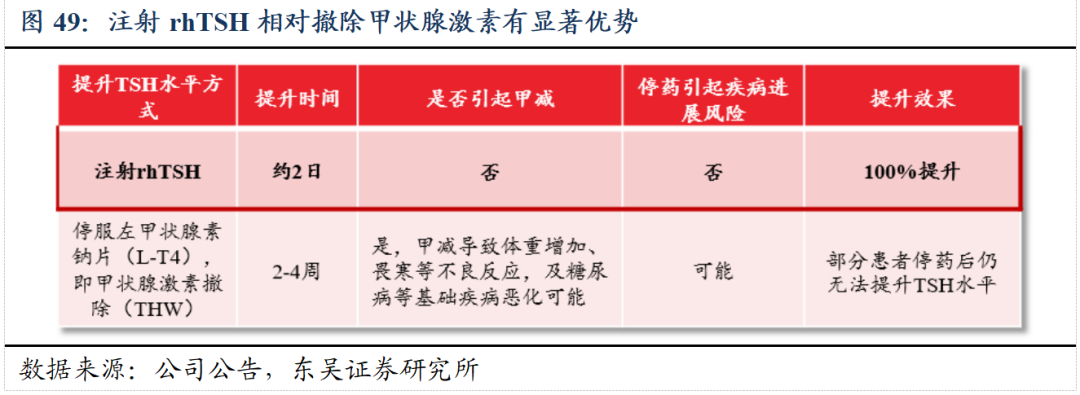
与德国默克达成独家商业化合作。与德国默克达成注射用重组人促甲状腺激素(rhTSH)在国内的独家商业化合作,公司将获得授权款总金额为人民币2.5亿元,同时公司将根据协议约定按净销售额两位数百分比支付市场推广服务费。默克在国内深耕甲状腺疾病领域多年,拥有成熟的商业化团队和成功的推广经验,待获批后将积极开展市场教育及准入工作。
5.3. ZGGS18:作用机制独特,增强肿瘤免疫抗肿瘤疗效
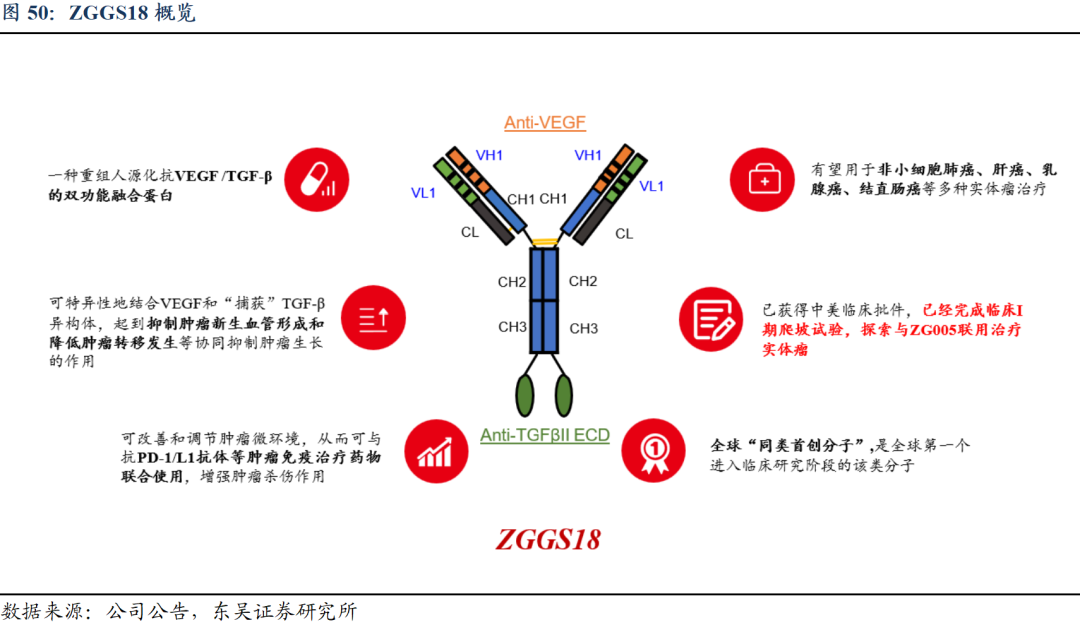
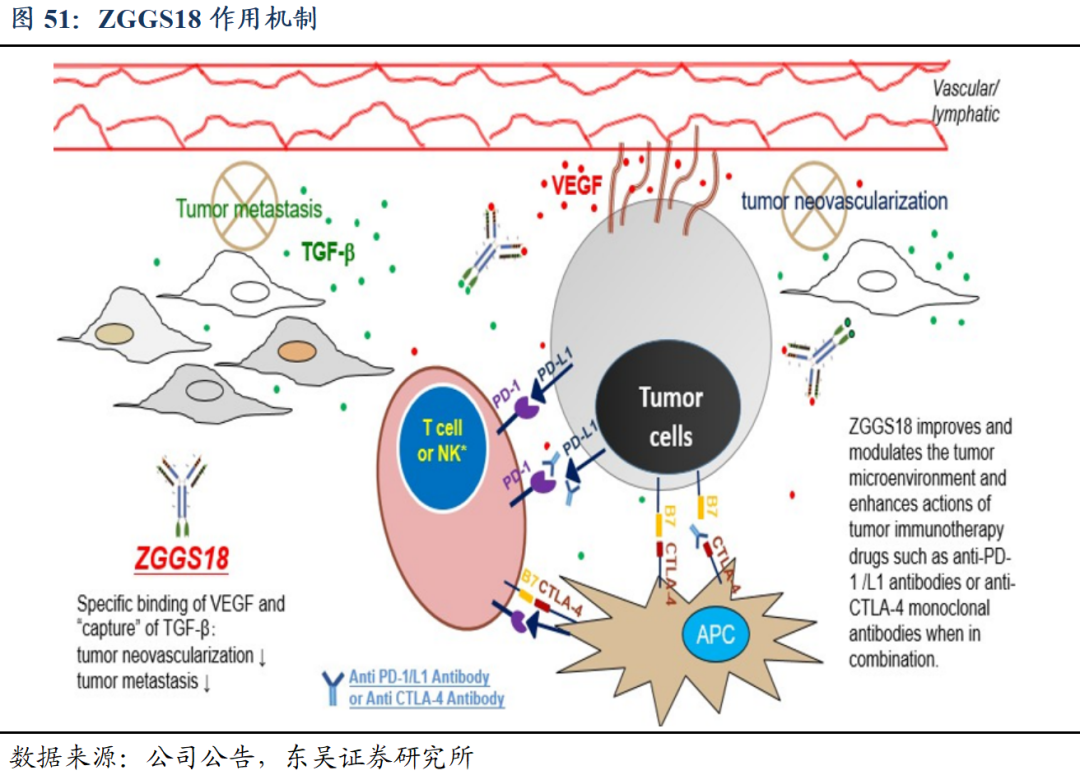
独特的作用机制:改善肿瘤微环境,增强肿瘤免疫抗肿瘤疗效。ZGGS18的双靶点作用能够同时减少肿瘤微环境中的VEGF和/或TGF-β,可以恢复肿瘤对肿瘤特异性细胞疗法的敏感性,并可能改善抗PD-1/ZG005/抗CTLA-4治疗的疗效。
1)阻断VEGF信号通路:①选择性与VEGF结合并阻断其生物学活性,减弱或阻止VEGF与VEGFR结合,以减少肿瘤血管形成,抑制肿瘤的生长。②使肿瘤血管正常化,改善血管通透性,增加肿瘤组织药物浓度,发挥抗肿瘤作用。
2)阻断TGF-β信号通路:①减轻Treg介导的免疫抑制,促进T细胞向肿瘤中心的渗透,从而引起强烈的抗肿瘤免疫和肿瘤消退。②增强CD8+T和自然杀伤(NK)细胞的活性,促进IL-2依赖性生长和淋巴因子激活的杀伤细胞(LAK)的分化。③增强肿瘤对免疫检查点抑制剂(ICI)的反应,如抗程序性死亡受体1(PD-1)或PD-1配体(PD-L1)的抗体。
5.4. ZGGS15:LAG-3/TIGIT双抗,有望治疗多种实体瘤
ZGGS15为1类创新型肿瘤免疫治疗生物制品,是全球首款进入临床研究的LAG-3/TIGIT双特异性靶点药物,目前全球范围内尚未有同类机制药物获批上市。
ZGGS15拥有双靶向阻断LAG-3和TIGIT的作用,既可以通过有效阻断LAG-3与其配体MHC-II等的信号通路,又可以有效阻断TIGIT与其配体PVR等的信号通路,促使PVR结合CD226产生共刺激激活信号,进而促进T细胞和NK细胞的活化和增殖,并产生细胞因子,从而具有协同增强免疫系统杀伤肿瘤细胞的能力。
2025ASCO披露:ZGGS15在晚期实体瘤患者中的剂量递增、耐受性、安全性、药代动力学的I期临床研究(ZGGS15-001)数据及最新进展:单药呈现出良好的耐受性和安全性以及抗肿瘤疗效,并有望与其它抗肿瘤疗法(如PD-1或PD-L1抑制剂等)联合治疗,起到良好的协同增效抗肿瘤作用。
截至2025年1月8日,共入组22例患者,其中 11例(50.0%)既往接受过至少3线系统治疗,8例(36.4%)既往接受过PD-1或PD-L1抑制剂治疗。①有效性方面,17例可评估患者中,6例达到SD,DCR为35.3%。在8例肺腺癌患者亚组中,5例(62.5%)达到SD,包括2例既往接受过≥2线系统治疗并维持SD超过36周的患者。②安全性方面,从0.3 mg/kg到30 mg/kg的剂量递增研究中未观察到DLT事件。90.1%患者发生了TRAE,其中仅1例患者出现淋巴细胞计数减少的3级TRAE,未见4级或5级TRAE,未见导致剂量降低的TRAE。
5.5 ZG2001:泛KRAS抑制剂,具有治疗多种KRAS突变肿瘤潜力
抑制机制:选择性结合SOS1的催化位点,阻止SOS1和非活化的GDP-KRAS结合,抑制GDP-KRAS转变为活化的GTP-KRAS;从而阻断了MAPK下游通路,引起肿瘤凋亡和肿瘤消退。
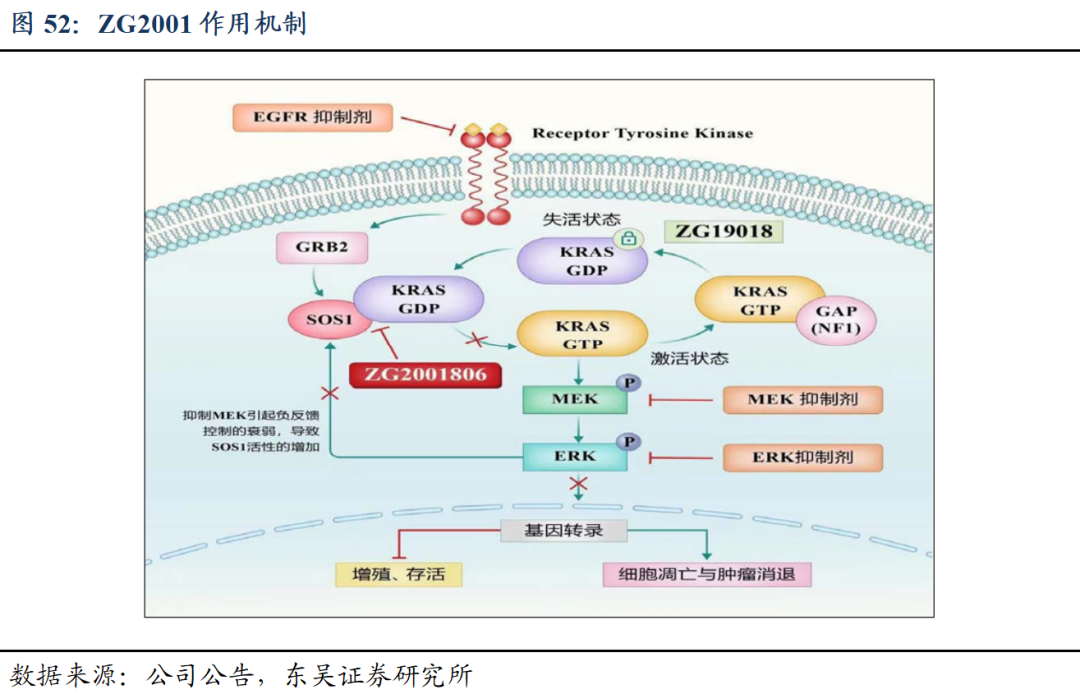
ZG2001是全球临床研究进展最快的SOS1选择性抑制剂之一,中国和美国已经获批临床,并正在I/II期临床研究中,具有独特的化合物结构,与现有临床化合物结构差异大,相对于其它临床药物分子,ZG2001具有更高的选择性和更好的体外抗肿瘤增殖作用。体外及体内药效学实表明,ZG2001可以剂量依赖性地抑制不同KRAS突变的肿瘤细胞增殖。
ZG2001具备良好的药代动力学特性,血浆暴露量高、组织分布广泛,在不同种属中都有类似的PK性质,没有种属特异性变化;此外与KRAS-G12Ci、KRAS-G12Dimulti-KRASi、MEKi、EGFRi、化疗、大分子药物等联用增效抗肿瘤作用。
6.盈利预测与估值
1)销售预测
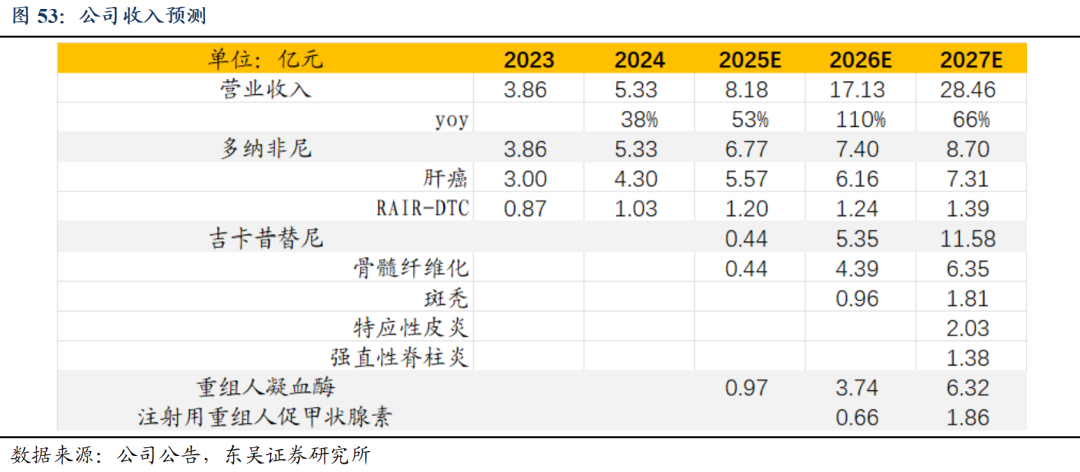
1)多纳非尼:多纳非尼为目前针对肝癌一线治疗和晚期分化型甲状腺癌(RAIR-DTC)治疗的核心药物,已上市2-3年,预计多纳非尼整体销售峰值可达10亿,预计2025-2027年收入6.77、7.40、8.70亿元。
2)吉卡昔替尼:骨髓纤维化(MF)适应症已获批上市,斑秃适应症已披露三期临床数据较优,考虑MF纳入医保后快速放量,中重度AD及AS适应症处于三期临床,2027年有望获批上市,预计吉卡昔替尼MF适应症加斑秃适应症销售峰值可达20亿,预计2025-2027年吉卡昔替尼整体收入(含自免)0.44、5.35、11.58亿元。
3)重组人凝血酶:作为国内首家上市的重组人凝血酶产品,手术刚需性强,考虑2024年底进入医保后快速放量,预计整体销售峰值可达20亿,预计2025-2027年收入0.97、3.74、6.32亿元。
4)注射用重组人促甲状腺素:2025年底分化型甲状腺癌术后诊断的适应症有望获批上市,考虑合作伙伴默克销售能力突出,预计整体销售峰值可达6-8亿,预计2026-2027年收入0.66、1.86亿元。
2)毛利率预测:考虑公司2025年为核心管线商业化关键阶段,2026年开始销售迅速放量,销售成本增加,预计2025-2027年毛利率为88%、85%、75%。
3)费用预测:由于公司2025-2027年进入关键三期药物较多,研发费用显著提升,预计2025-2027年为5.00/6.14/6.81亿元;同时初上市药物也较多,销售费用有所提升,预计2025-2027年为2.82/3.41/4.54亿元;管理费用率在4-8%左右。
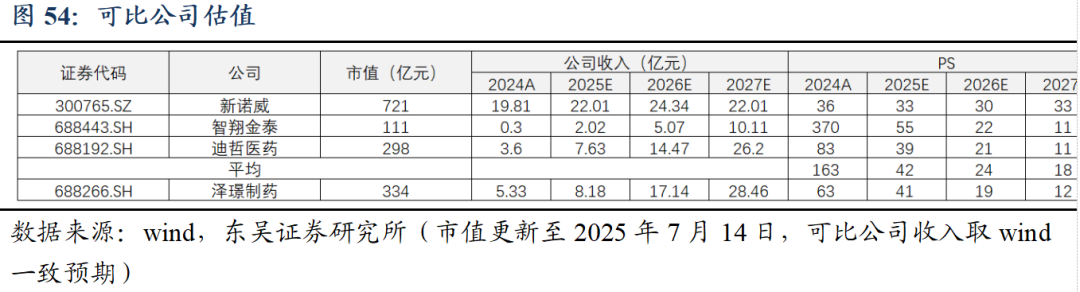
综上,我们预计公司2025-2027年收入分别为8.18/17.14/28.46亿元,归母净利润分别为-1.00/3.77/7.71亿元。考虑公司目前处于创新商业化兑现初期,部分核心管线上市时间较短,国内销售初步放量,我们选取PS相对估值法。
我们选取管线结构相近、盈利阶段相近的新诺威、智翔金泰、迪哲医药作为可比公司,泽璟制药2025-2027年PS计算得到41/19/12倍,可比公司对应平均42/24/18倍。我们认为泽璟制药目前处于创新管线兑现早期,产品快速放量在即,且体内在研管线层次丰富,ZG005、ZG006均具备成为BIC药物的潜力,公司正由Biotech迅速转型为Biopharma,首次覆盖,给予“买入”评级。
7.风险提示
1、新药研发进展不及预期风险
公司基于未来发展所需,每年投入大量资金用于药品的研发,此外未来核心产品ZG005、ZG006开展Ⅲ期临床时需要针对海内外病人投入大量研发成本。随着国家监管法规、注册法规的日益严格,药品研发存在不达预期以及药品注册周期可能延长的风险。
2、药品销售不及预期风险
公司近两年商业化产品吉卡昔替尼、重组人凝血酶随着纳入国家医保目录,有望加速放量。但若药品销售不及预期,则会对公司现金流产生影响,可能导致盈亏平衡期延长。
3、海外交易不及预期风险
公司核心在研产品有海外授权的可能,若海外交易谈判失败或交易金额不及预期,则会对公司部分产品价值认知产生不利影响。