����Ͷ��Ҫ��
����Ͷ��Ҫ��
����Ͷ��Ҫ�㣺��˾�������й���1��TYK2���Ƽ�Ϊͬ���Ʒ��BIC��������չ�����������Ӧ֢��������BDDZ����2���ڷ�SERD���ESR1ͻ���ER+���ٰ���Ч����ȫ�Ը��ţ������ڹ���ͬ�ྺƷ������ʵ����ҵ�������������в�Ʒ��������&���������۽���Сϸ���ΰ����뱴��ҩҵ&��������������ۣ��ͷ���ҵ��DZ����
����D-2570����ѡ����TYK2�乹���Ƽ���PSO��PsA��UC������Ӧ��ǰ����������������г�����˾D-2570��Ϊ�ڷ���ѡ����TYK2���Ƽ�����乹���ƿ�����������JAK������صĸ�Ⱦ�ȷ��գ���м�����ڵ�12��PASI75��90%����PASI100��50%��Ӧ���ʾ���������TYK2���Ƽ������ˮƽ��������������ٴ�Ԥ��26H2��ùؼ����ݣ���������ѻ�FDA�����������ݽ�����̽��ȫ������ٴ������⣬D-2570��Ӧ֢����չ��SLE��PsA��UC������UC��Ӧ֢�ٴ�������25M5���飬Ԥ��26Q3��ä�������ݣ�ä̬�¹۲쵽��ȫ�����������ȫ�Է���Ԥ�ڣ�������ΪIBD����ͻ�����Ʒ���������Ϊ�÷���������Ϊ���Ƕ����⼲���ס���BICʵ���Ŀڷ���Ʒ���������亣��BDDZ����
����D-0502���ڷ�SERD�����ٰ����ٴ���ֵ�ԣ���ҩ�Ϸ�ά˾Ⱥչ�ָ��Ű�ȫ�Լ�������ǰ���� ��˾D-0502��Ϊ��һ���ڷ�SERD�����ESR1ͻ���ER+/HER2-���ٰ�����չ�ָ�����Ч�����й��г�Ψһ���е�SERD����ҩ��ά˾Ⱥ��ȣ�D-0502��ORRΪ12.5%��VS9.1%����CBRΪ50%��VS45.6%����mPFSΪ7.4���£�VS6.5���£����ڻ������������չ�ֳ����õİ�ȫ�ԺͿ���������ǰ����ĿǰD-0502���ڹ��ڢ��ڣ�������Ϊ�������ڹ����ڷ�SERD������ʵ����ҵ����
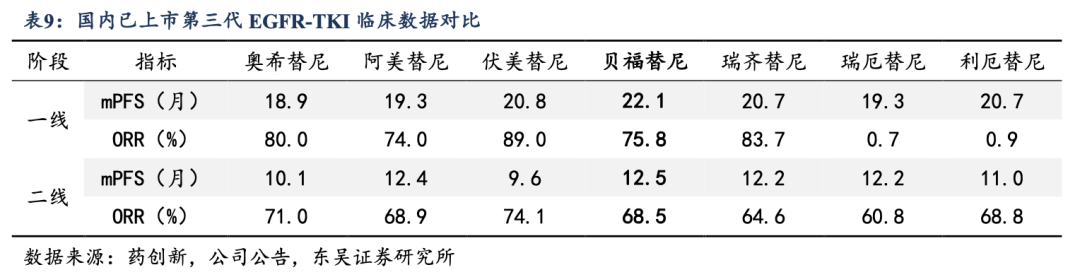
�����۽���Сϸ���ΰ��������Ʒ��������&���������뱴��ҩҵ&��������������ۣ��ͷ���ҵ��DZ������������Ϊ��˾���е�����EGFR-TKI����һ��NSCLC��2024�����ҽ����һ�ߡ�������ҩ������mPFS�ֱ�Ϊ22.1%��12.5%��Ϊͬ�ྺƷ���ţ��Ҳ�Ʒ�뱴��ҩҵ������δ���������ٷ�������������Ϊ���ڵڶ������е�KRAS G12C���Ƽ�����mPFS��9.1���£���ORR��52%�������ͬ�ڻ�����Ʒ�൱����mOS��14.1���£��������������о�Ʒ���÷���δ��������չ��NSCLCһ�����Ƽ���ֱ�����������Ƶ�����Ŀǰ��������������������������۷ֳɡ�
����ӯ��Ԥ����Ͷ������������Ԥ�ƹ�˾2026-2028������ֱ�Ϊ1.09/6.03/9.51��Ԫ��������D-2570��DZ��BD�������ǹ�˾ӯ���ݲ��ȶ�������DCF��ֵ��������Ŀ��۸�39.03Ԫ�����⣬��˾��������D-0120��ʹ�磩�ȶ���������õ�ҩ������ݶȻ����������������������������ҵ����������DZ�����ߣ��״θ��ǣ����衰���롱������
����������ʾ���ٴ����Ȳ���Ԥ�ڣ������������Ԥ�ڣ���ҵ������Ԥ�ڵȡ�



����1. �淽���������������⼰��л�����³ɹ�����ת��
����1.1. רע���������⼰��л�����߹ܼ��з��Ŷ�רҵ�������
�����淽���������2013�꣬�ܲ�λ���Ϻ��Ž�����˾�۽����������⼰��л���ش����������в�Ʒ��Ϊ�����з���ӵ������ȫ��֪ʶ��Ȩ����˾�ѳɹ������������ȵ�С����ҩ���з���ϵ������һվʽ���з�ƽ̨�������˴Ӱе�ɸѡ����֤�����������ҩ����ơ�ҩ����ת��ҽѧ������ѧ�ϳɹ��պ��ٴ�������ȫ���̡����⣬��˾�����˼��������ҩ�����ƽ̨�����ǵ�����С���ӵĹ����������ʽṹǶ��ʽģ�⡢��ά��״ƥ��Ͷ�ά�����Ǻ���Ƶ��������������˷�����Ƶľ��Ⱥ�Ч�ʡ�
�����߹ܼ��з��Ŷ�רҵ���������ҩ�з�������ḻ����˾�ĸ������㼰�����з��Ŷ��ɶ�λ���ڹ��ʶ�����ҩ��˾���θ��з�����ְ�������ר����ɣ�ƽ��ӵ�г���20�����ҩ�з����顣������ҩ��ѧ�����ͷḻ���з���������Ϊ��˾�Ĵ��·�չ�춨�˼�ʵ�����������Ŷӳ�Ա��С����ҩ����ơ��ٴ��о����Ŷӹ����ȷ�����߱��ܳ���רҵ�����ͳɹ�����Ŀ���顣

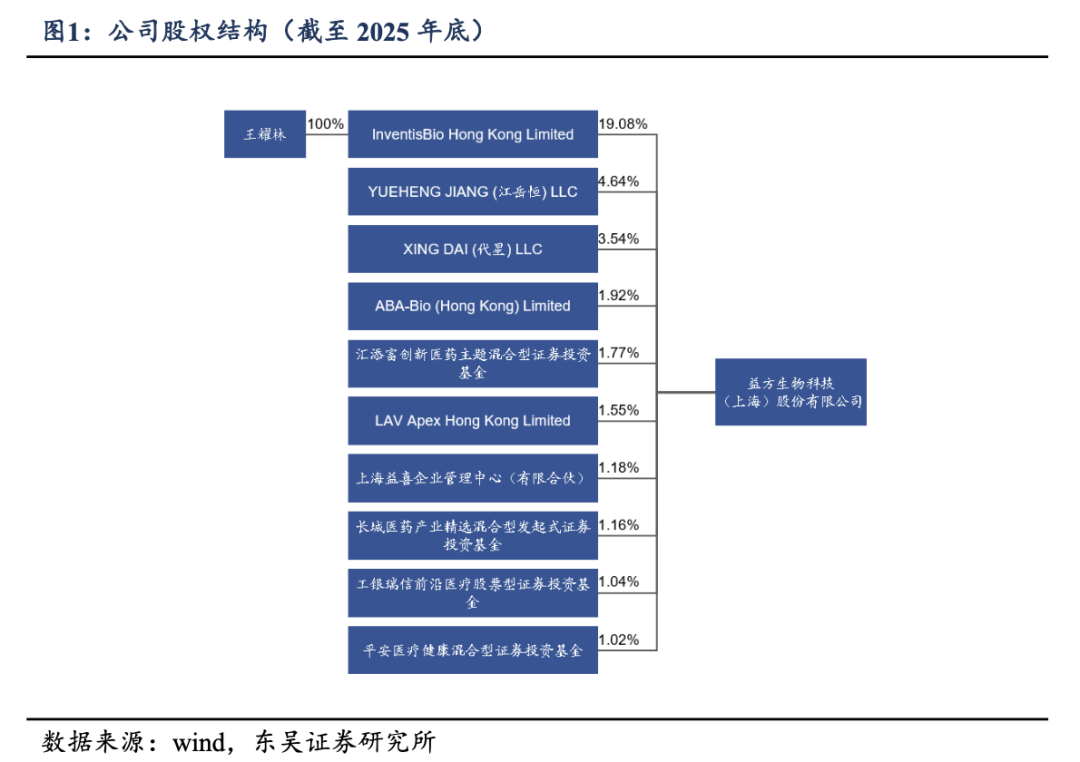
������Ȩ�ṹ������ʵ����Ϊ��λ���ϴ�ʼ�ˡ���˾�Ĺ�Ȩ�ṹ�������ȶ���ʵ�ʿ�����Ϊ��ҫ�ֲ�ʿ�������㲩ʿ�����Dz�ʿ��λ���ϴ�ʼ�ˡ�����2025��ף�����ͨ������淽��InventisBio Hong Kong Limited����YUEHENG JIANG LLC��XING DAI LLC��ͬ���ƹ�˾���ֱ�ֹ�19.08%��4.64%��3.54%��ȷ���˹�˾��չս�Եij���һ���ԡ�
����1.2. �ֽ�����ã��з��˳���Ͷ�룬���³ɹ�ת������
�������ù�˾�������ᡢ�����������ٷ��������Ĺ�����������ҵ������˾Ӫ����ҪԴ�ڼ�����Ȩ�ͼ���������2023-2025��ֱ�Ϊ1.86��1.69��0.37��Ԫ�������벨���ϴ�ԭ��ϵ��Ȩ�ͼ�����������Ĺ����ڲ�ͬ��ȴ���һ�����졣������Ϊ�����ű���ҩҵ���������ר�����ڡ��Լ�������������KRAS G12Cͻ����NSCLC��Ӧ֢������2026�꿪ʼ����ҽ����������˾����������۲�Ʒ�����������۷ֳɣ�ͬʱ��˾���Ĺ���D-2570��D-0502����������ҵ������˾������ʵ��ӯ��ƽ�⡣
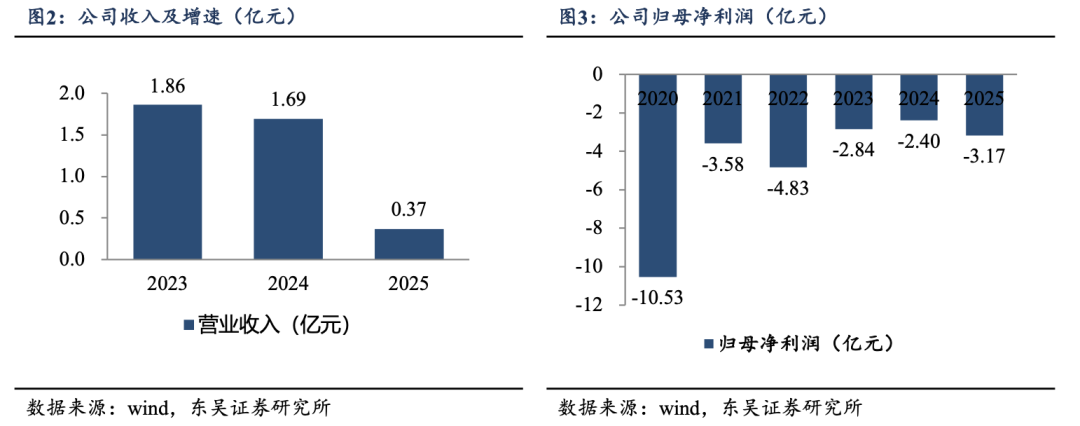
�����ֽ���״����ã���ҵ�����̻����ƽ�����˾��Ӫ���ֽ�����2022���-4.77��Ԫ���ƣ�2025��Ϊ-1.55��Ԫ�����Ŷ�����Ȩ��Ʒ����̱������δ�����۷ֳɵ����֣���˾�ֽ���״�������õ��������ơ���2023��8���������������Ȩ����Ϊ�����������罫��˾֧����߲�����5.5��Ԫ�������̱�������꾻���۶�ֲ�֧������Ȩʹ�÷ѣ�Ϊ��˾δ�����ֽ����ṩ����֧�š�
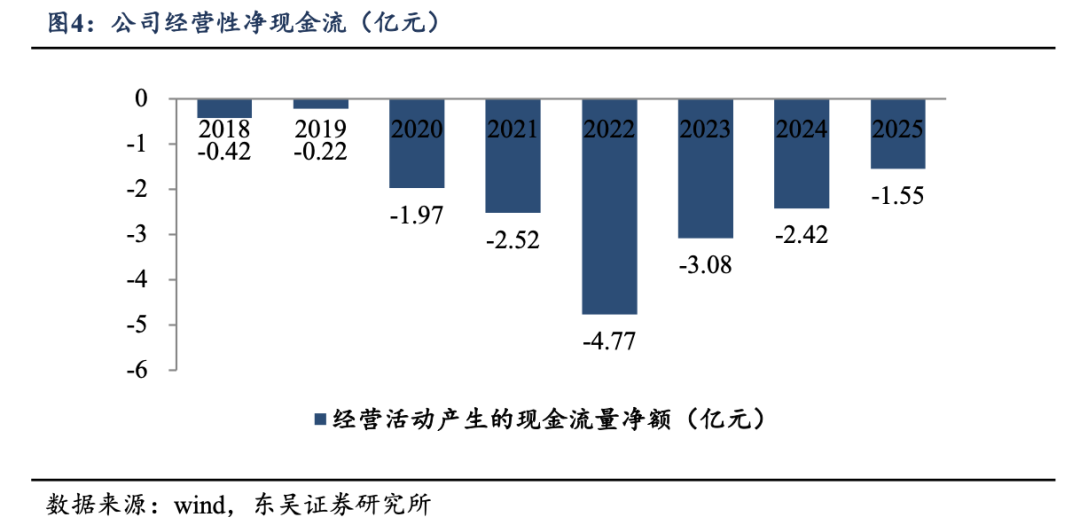
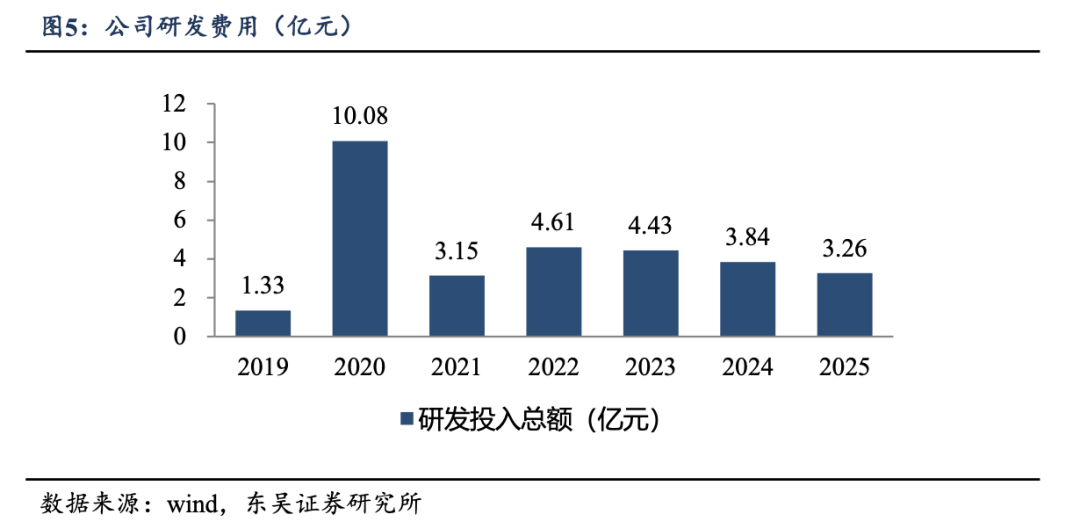
�����з��˳���Ͷ�룬���ٴ��³ɹ�ת������˾ʼ�ձ����Ÿ�ǿ�ȵ��з�Ͷ�룬2021-2025��ֱ�Ϊ3.15��4.61��4.43��3.84��3.26��Ԫ��������Ͷ��Ϊ���߲�Ʒ�Ŀ����ƽ���δ����Զ��չ�ṩ���������ϡ�Ԥ��δ�������з����߳����ƽ�����˾���������ֽϸ�ˮƽ���з�Ͷ�롣
����1.3. ���й��߷ḻ����ҵ����ֵ������֤�����ڷ�չ������
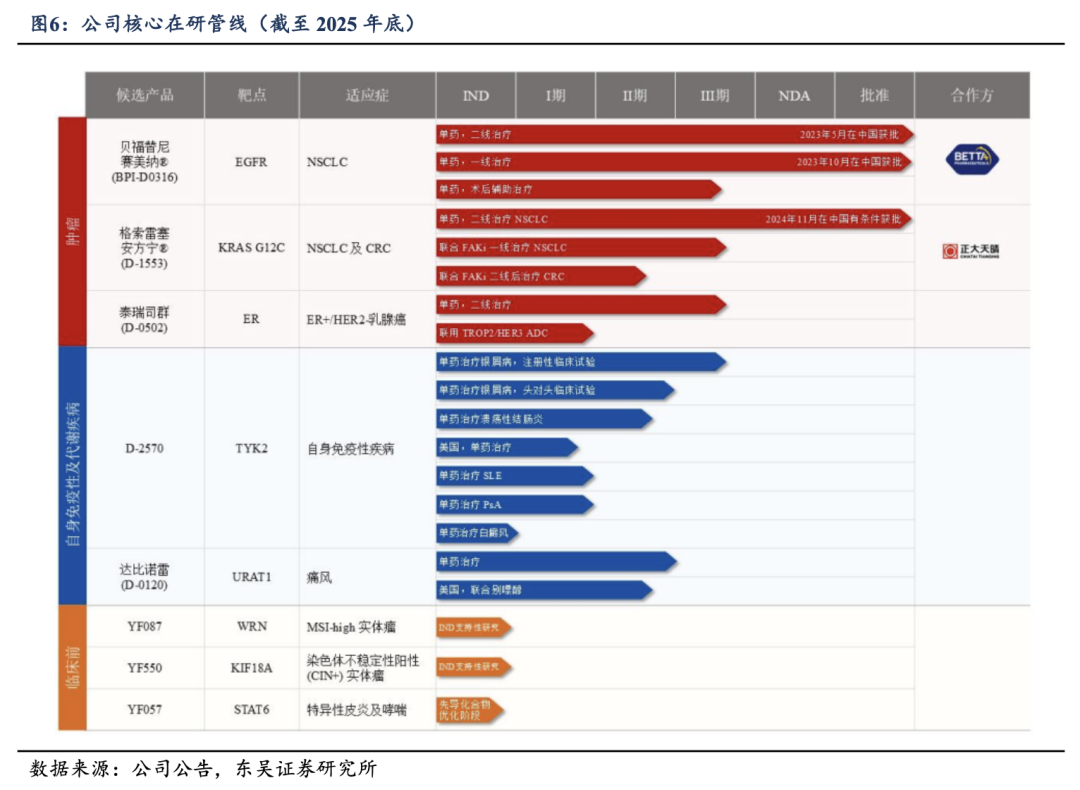
������˾���й��߷ḻ���з���������ѭ����Ϊ���ڷ�չע��ǿ������������2025 ��ף���˾���ж���Ʒ���ڲ�ͬ���з��Σ��������������⼰��л�༲���ȶ����������˾�ѳɹ��ƶ����������з��Ĵ���ҩͨ��������Ȩ������ģʽ�������У��ֱ����뱴��ҩҵ�����ĵ�����EGFR���Ƽ���������BPI-D0316���Լ����������������KRAS G12C���Ƽ���������D-1553�������Ʒ��������ҽ������־�Ź�˾���з��ɹ��ѳɹ���ҵ�����֣������ջ��ڡ�����2����IJ�Ʒ��TYK2�乹���Ƽ�D-2570���ڷ�ѡ���ԴƼ������彵���D-0502���ѽ���III��ע�����ٴ�����Σ��������Ʒ���ڢ��ڻ�����ٴ�����Ρ����⣬��˾������3������ٴ�ǰ��Ŀ�������ḻ��չ�ֳ���˾�Ӱе㷢�ֵ���ҵ�����ǿ��ȫ��ҵ���з���ת��ʵ����
����2. ���Ĺ����Ʋ��컯���ٴ�����ٶ�������
����2.1. D-2570����ѡ����TYK2�乹���Ƽ�����������г�
����2.1.1. ����TYK2���������������֢ͨ·��PSO��PsA��UC������Ӧ��ǰ������
����TYK2��ΪJAK��ø�����Ա�����źŴ���ͨ·���нϸߵ������ԣ���Ҫ����IL-12��IL-23�Լ�I�����صȹؼ�����ϸ�����ӵ������ź�ת������㷺����JAK1-3�Ĵ�ͳ���Ƽ���ȣ�ѡ������TYK2������ͨ���乹��ʽ�����ټ�ø�ṹ��JH2�����ܹ���������������Լ����ĺ�����֢ͨ·����һ�����������Ͽ������������봫ͳJAK������ص����ظ�Ⱦ��Ѫ˨�γɵȷ��գ��Ӷ�Ϊʵ�ָ��Ż������Ʒ��ջ���ȵ춨�˿�ѧ������
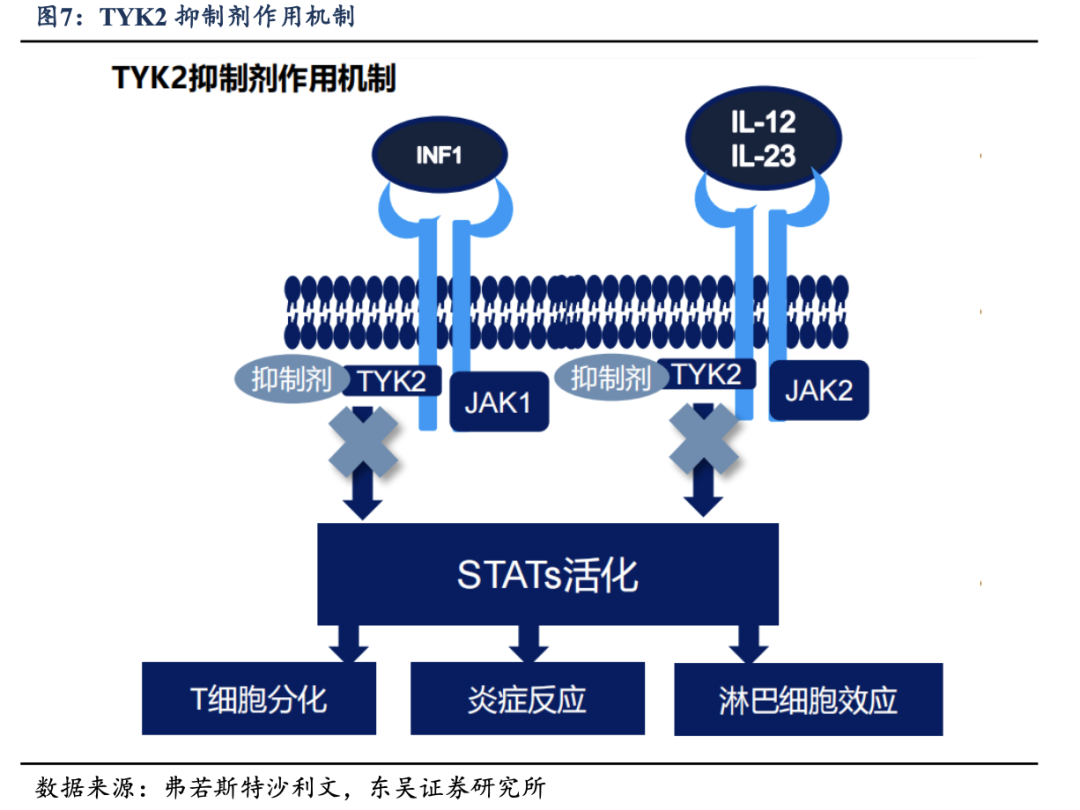
�������������������ƣ�TYK2���Ƽ��ڶ���ش�����������Լ�������չ�ֳ�������Ӧ��ǰ�����������Ӧ֢�������ضȰ߿�״��м����PSO������м���ؽ��ף�PsA���Լ������Խ᳦�ף�UC���ȡ����У�TYK2���Ƽ����PSO����Ч��Ϊͻ��������ԭ��ΪTYK2��IL-23�ź�ͨ·�еĺ��ĵ�λ��IL-23ͨ������TYK2/JAK2������崫���źţ�����TH17ϸ���ķֻ��������������������ЧӦϸ������IL-17�����ź���Ϊ��м����֢��·�ĺ����������ء���ˣ�����TYK2�ɴ����θ�Ч��ϸ�ͨ·�������ε�IL-17ˮƽ����Ϊ�ؼ��������־��������ٴ�������ֱ������TYK2���Ƽ���ͨ·����Ч�����ҩDZ����
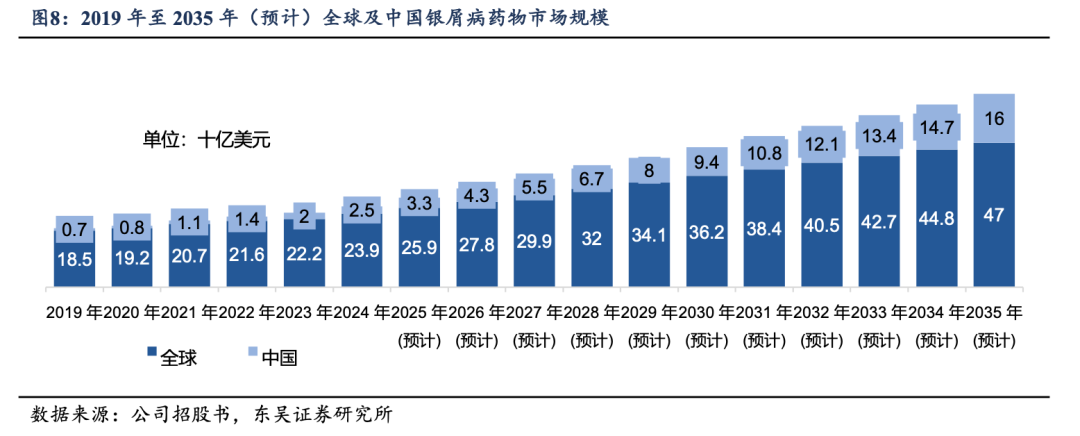
�����߿�״��м����һ��������ϵͳ�쳣�鵼��������֢��Ƥ�������ٴ�����Ƥ�����ָ�������ɫ��м�ĺ�ɫ�߿�Ϊ��Ҫ�������������ⲿ��ϥ����ͷƤ�Ȳ�λ��PSO�������������Գ����Σ��Ի��ߵ�����������ɳ���Ӱ�졣���ݸ���˹��ɳ�������ݣ�2024��ȫ���������Ѵﵽ1.73������Ԥ�Ƶ�2030�꽫����1.81�������й��г����棬2024�껼������Ϊ590���������ȶ����������е����������Ƽ���Ϊע���ҩ���ڳ��������б���Բ��㣬����ͳ�ڷ�ҩ������Ч�밲ȫ�����������ھ��ޡ���ˣ���߿ڷ������Ժ;��������û��Ƶ�TYK2���Ƽ���ǡ������ٴ����ƵĿհף����ƶ�������м��ҩ���г��еĿ�������
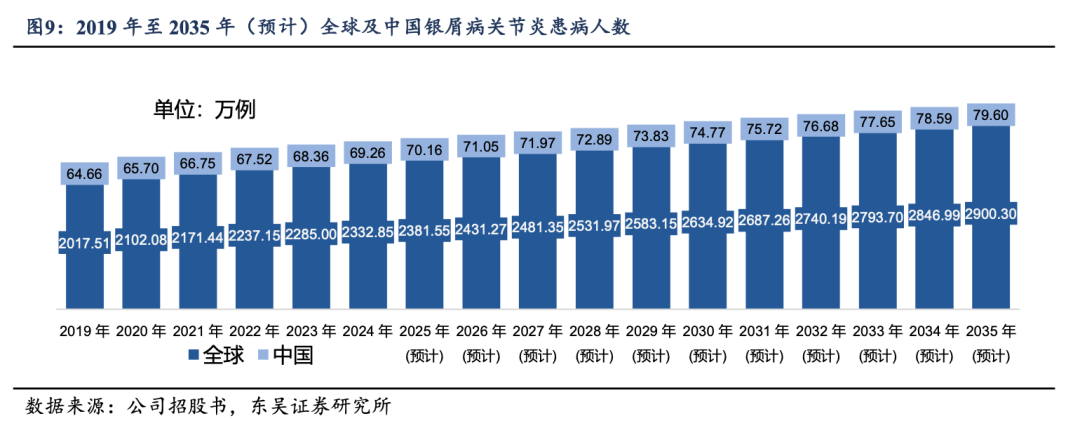
������м���ؽ�����һ������м����ص���֢�Թؽڲ����ɷ����ڲ�����м�������У��䲻�����Ƥ�������ᵼ�¹ؽ���ʹ���������������Ի��Σ�������Ϻ�Ԥ������Ҫ��PsA�Ļ��߹�ģ��С��PSO�����������Ƹ�Ϊ���ԡ��ݸ���˹��ɳ�������ݣ�2024��ȫ��������ԼΪ2333������Ԥ�Ƶ�2030�꽫������2635����������ͬʱ����Ƥ���ؽ�֢״���ŵĹ������ߣ��ٴ�������Ҫ�ܹ�һ�廯�����ķ�����TYK2���Ƽ�������һ�ֿڷ�ҩ��ͬʱ�������ֲ��䣬�����Ƶ�ͬʱ���������ԡ�
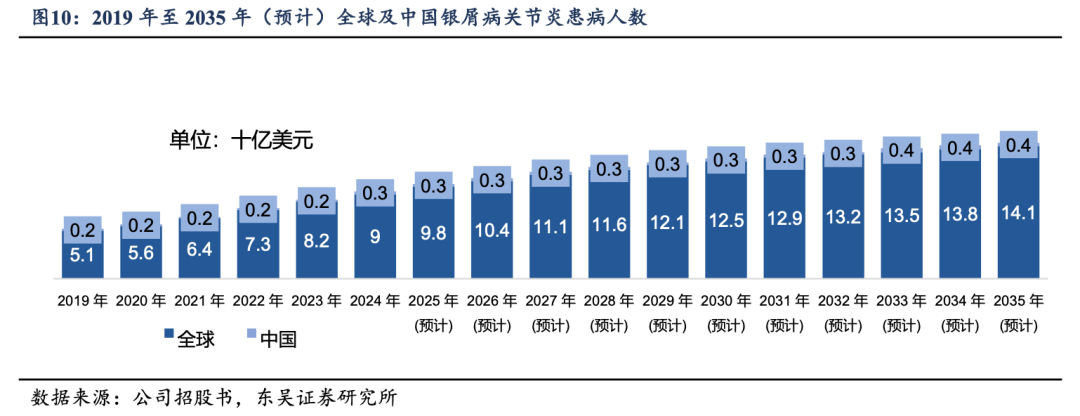
���������Խ᳦����һ����Ҫ�ۼ��᳦�Ĥ�����ԡ���������֢�Գ������ٴ�����Ϊ�����������ĸ�к���ҺŧѪ���ʹ���䲡�̳ʻ���뻺���ڽ��棬����Ŀ�������յ���ά�ֻ��⡣UC����Ⱥ������ȫ��Χ��Ѹ�����ݸ���˹��ɳ�������ݣ�2024��ȫ�������Ѵ�500������Ԥ�Ƶ�2030�꽫����690�����������й����ߵ������ٶ���������ȫ��ˮƽ�������һЩ��ͳ�������Ƽ�����ѡ���Ե�TYK2���Ƽ������Ͼ߱����ŵİ�ȫ��DZ���������ϻ��߶Գ���ά�����Ƶİ�ȫ��Ҫ��
�������⣬ϵͳ�Ժ���Ǵ���SLE��������Ҳֱ��/�����TYK2���û�����ء�1��SLE�ɵ������ٳ�����֢��Ӧ����֯���ˣ�����ѪҺ�л���ڹ�������������֯���쳣���壬�Ǹ߷����ʺ���������ص����⼲�������ݸ���˹��ɳ���ģ�ȫ��SLEҩ���г���ģ�Ѵ�2019���14����Ԫ������2024���34����Ԫ��Ԥ��2035���208����Ԫ��2�����综�ߵ�����ϵͳ�ṥ��Ƥ���еĺ���ϸ��������ɫ���쳣������������ͷ������ë��üë���ݽ������ݸ���˹��ɳ���ģ�2024��ȫ��/�ҹ����综�߷ֱ�Ϊ6180��/705������Ԥ�Ƶ�2035��ֱ��6800��/730������
����2.1.2. D-2570�����ض�PSO����չ��BICDZ����ȫ���ٴ�����Ӧ֢��չ��Ч�ƽ�
����D-2570���淽���������з���һ��ڷ�����ѡ����TYK2�乹���Ƽ����ٴ�ǰ������ʾ���TYK2��ѡ����Զ����JAK1��Ԥʾ��DZ�ڸ��������ư�ȫ�������з����߲���������D-2570�����ض�PSO��չ��ΪѸ�٣�Ŀǰ�ѽ���III���ٴ���ͬʱ�����UC��PsA��SLE����Ӧ֢Ҳ�ѽ�������ٴ���
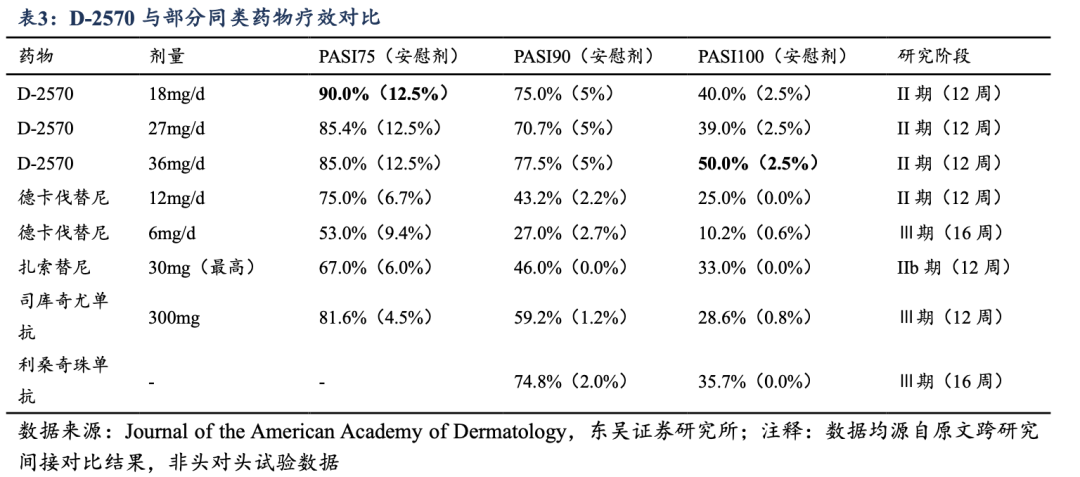
����D-2570�����ض�PSO����������չ��BICDZ��������Ա���Ҫ����TYK2���Ƽ�����м��II�����ݣ�D-2570��12��PASI75��90%����PASI100��50%��Ӧ���ʾ��������ˮƽ����ȷȷ�������ڿڷ�TYK2���Ƽ��е���Ч����
��������D-2570��м��II���ٴ��о��������˾����չ���ڶ�����������Լ�������������ٴ�̽������ȫ���ٴ������ƻ�����Ч�ƽ�������2026��4����Ѯ��D-2570��м����Ӧ֢ע����III���ٴ���������2026��3�����ȫ�����������飻D-2570������м��ͷ��ͷIII���ٴ��������ڽ����У�D-2570��������ҩ������м����II���ٴ���������ʽ�������FDA����D-2570���������Խ᳦�ף�UC��II���ٴ����顢������м���ؽ��ף�PsA����II���ٴ����顢����ϵͳ�Ժ���Ǵ���SLE����II���ٴ�����Ҳ�����ƻ������С�
������ѧ�����棬D-2570�������ضȰ߿�״��м����II���о������2025��3���Կ�ͷ������ʽ������Ƥ����ѧ�ᣨAAD������Ϲ���������2025��12�·���������Ƥ����ѧ��Ĺٷ���ѧ���������Ƥ����ѧ����־����Journal of the American Academy of Dermatology��IF��11.79���������ʾ����12�����ƺ�D-2570��18mg���ͼ�������27mg���м�������36mg����������ֱ���90.0%��85.4%��85.0%�Ļ��ߴﵽPASI75��Ƥ���������75%������Ҫ�յ㣬�������ڰ�ο���飨12.5%��p<0.001����ͬʱ��D-2570��PASI90��PASI100Ӧ���ʷ���Ҳ����ͻ����D-2570�͡��С��������PASI90Ӧ���ʷֱ�Ϊ75.0%��70.7%��77.5%��PASI100Ӧ���ʷֱ�Ϊ40.0%��39.0%��50.0%�����������ڰ�ο���飨5.0%��2.5%��p<0.001����D-2570����Ч�������������е�ͬ��TYK2���Ƽ���������Ч���Ͽ��뿹������ҩ���翹IL-17A,��IL-23���壩����������ȫ�Է��棬D-2570�������������ã�����������¼�Ϊ������жȣ�δ�������ز����¼����䰲ȫ������������TYK2���Ƽ����ƣ�δ�����µİ�ȫ���źš�
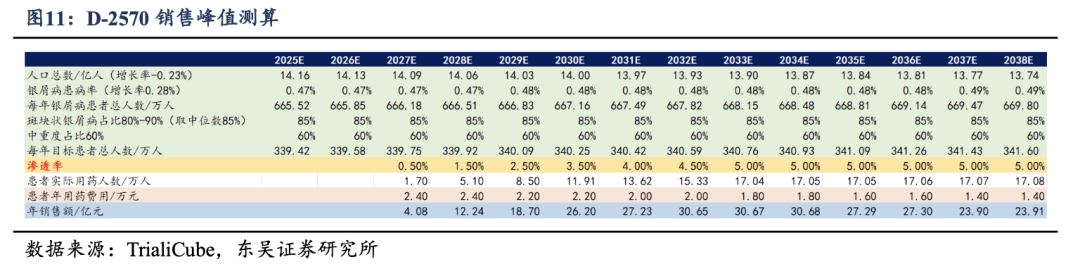
���������й����˿ڼ���м�������ʲ��㻼���������������ضȰ߿�״ռ��ȷ��Ŀ�껼�߳أ�����Ԥ��D-2570���۷�ֵΪ30��Ԫ������ؼ����������1���ֹ۹��Ʋ�Ʒ��2027��������У�2�������Ʒ��òο�ͬ��ҩ������趨Ϊ2.4��Ԫ��
����2.2. D-0502����һ���ڷ�SERD���з����ȹ�������
����2.2.1. ���ٰ�����Ⱥ���Ӵڷ�SERD�ٴ���ֵ��
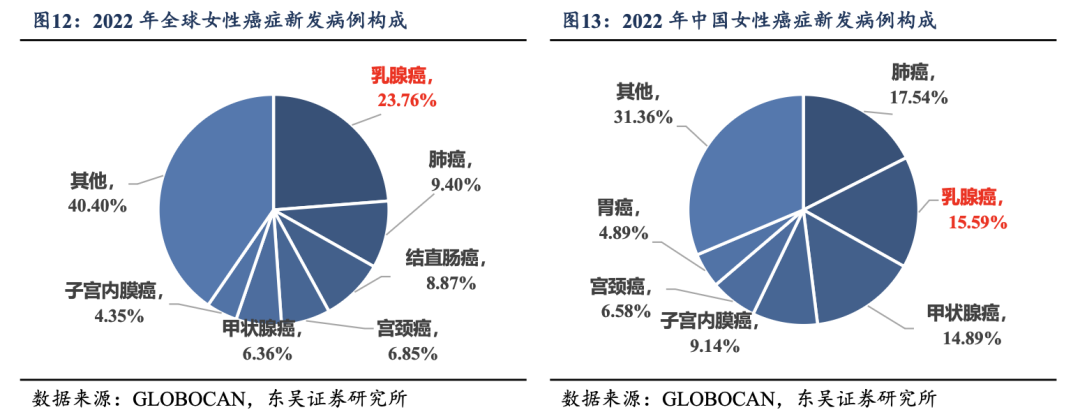
�������ٰ���Ϊȫ��߷����֣��������Ӵ���GLOBOCAN��ͳ�����ݣ���ȫ��Χ������2022�����ٰ���23.76%��ռ�ȳ�Ϊȫ��Ů��Ⱥ��������Ķ���������λ��Ů��֢�·�����������λ�����й������ٰ�ͬ������вŮ�Խ����ĺ��İ��֣�2022���й�Ů��֢�·�����35.7������ռ�ȴ�15.59%��Ϊ�й�Ů�Եڶ��߷�����������
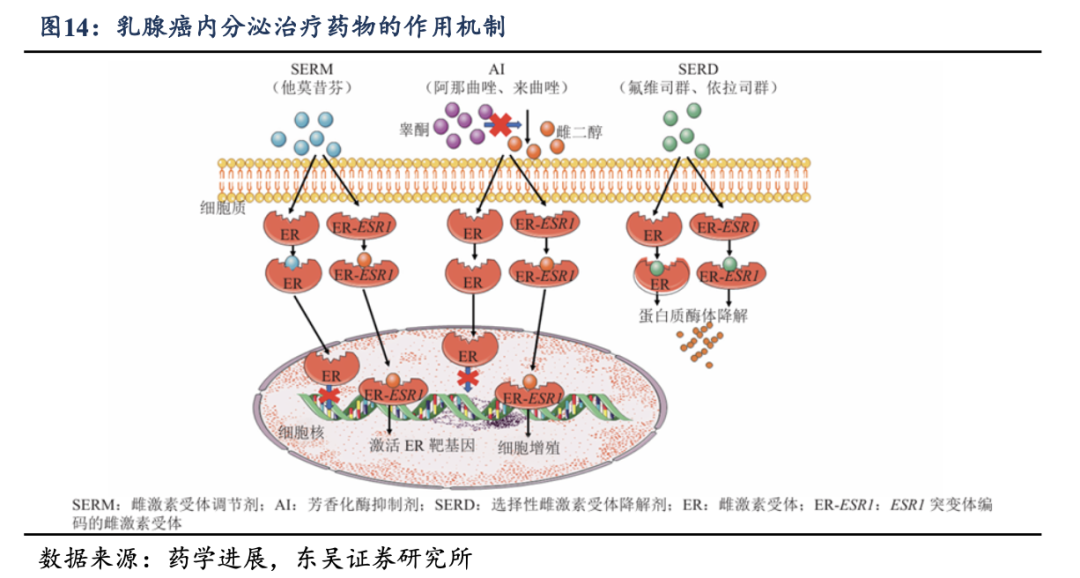
�����Ƽ������壨ER����HR+/HER2-���ٰ��ĺ��������е㣬��7�ɵ����ٰ�����ΪER���ԣ���鵼�ĴƼ����ź�ͨ·������ϸ����ֳ��ת�ƵĹؼ��������ڷ������Ƶĺ�������ͨ������ER�е���ϸ��ź�ͨ·����ͳ�Ʒ���SERM��ѡ���ԴƼ���������ڼ�������ϴƼ�����ER��ϣ���AI�����㻯ø���Ƽ����ɼ������ڴƼ��غϳɣ���Χ�ƴ˰е���ƣ�����ʵ�ֶе�ġ����ơ���ѡ���ԴƼ������彵�����SERD����Ϊ����ER����һ�����ԣ�ͨ������+���⡱��˫�����û���ͻ�ƴ�ͳ�ڷ����Ʒ��ľ��ޣ���Ϊ�ðе�����ŵ��ط���
������һ��SERD��ά˾Ⱥ����֤��ER������Ե��ٴ���ֵ�������������öȼ��ͣ�<1%���ҽ��ܼ���ע���ҩ������ER�е㱩¶�����㡢��ҩ�����Բ���������˰е��ֵ�ķ��ӣ��淽����D-0502��Ϊ��һ���ڷ�SERD��ͨ�����ӽṹ�Ż����ڱ���SERD����ER���Ļ��ƵĻ����ϣ�ʵ����ҩ������ѧ���ҩ��ʽ��˫��������ͬʱ��D-0502�����ESR1ͻ��ĴƼ�������ʱչ�ֳ����ŵ���Ч����һ���Ŵ��˰е���ٴ���ֵ��
�������й��г�����ά˾Ⱥ��ĿǰΨһ�������е�SERD�����ҩ���ɰ�˹�����з���2010����ʽ�����й��г���2017���������ҽ��Ŀ¼������Ħ��ҽҩ���ݿ⣬2019-2024���ά˾Ⱥȫ�����۶���������ƣ�2024���17.3��Ԫ��
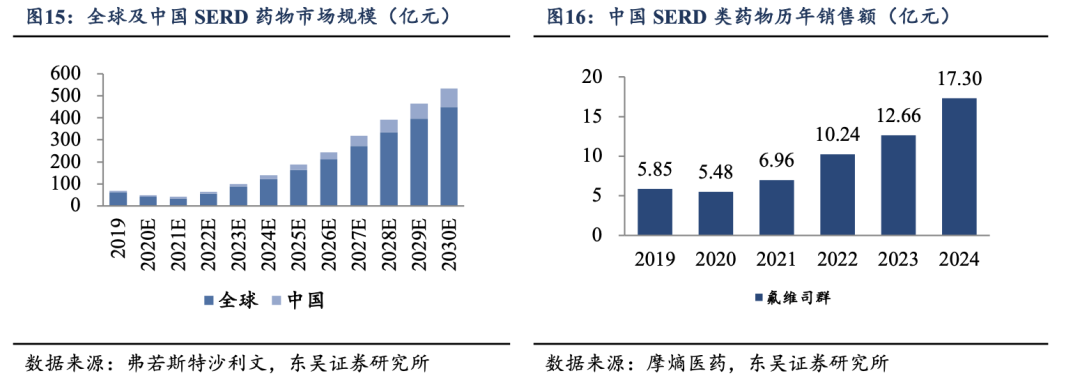
�������ٰ�����Ⱥ���Ӵ������ڷ������ƴ�������ȱ�ݣ����磺��Ī���ң�SERM�����ӹ���Ĥ������������ҩ����أ���������AI�������շ�ESR1����ͻ�䣬�����¹��ܶȽ��ͣ���FDA������˾Ⱥ��֤��SERD�����û��ƣ�ҲΪ����������ER���ԣ�ER+���������Լ������������ƣ�����ADC���ã����˿ռ䣬ͬʱ�ڷ�SERD�Ŀ������ע����ͣ��������������ҩ�����������ԡ�
��������2025��ף�������˾Ⱥ����ķ³˾Ⱥ����ڷ�SERD�ں��������У��й�����ҩƷ�ල�����֣�NMPA����δ���κ�һ��ڷ�SERD��ʽ���С��ڹ��ڿڷ�SERD���й����У���˾��D-0502�������ȣ��䵥ҩ���ڶ���HR+/HER2-���ٰ��Ģ����ٴ���2022��3��������
�����ڷ�SERDҩ�����ٴ������ձ�����з�ƿ������˾��Ʒ��ESR1ͻ����������ʾ�����ŵ���Ч���ݣ�PFSҪ��ȫ��Ⱥ����������˹�˾ѡ���˲��컯�ٴ����ԣ����Ⱦ۽���ESR1����ͻ�����ٰ�ϸ����Ⱥ�ƽ����У��Թ��ȫ��Ⱥ�����ĸ߷����볤���ڡ�ͬʱ��ͨ��̽���������DZ����ADC��ѡҩ������Ͽ���������ǿ���ڶ�����������ľ�������ʵ�֡����ڿ������С�����ȫ����չ������ռϸ�������ȷ����ƣ���Ϊ������ȫ��Ⱥ�г���չ�춨������
����2.2.2. D-0502����ҩ�Ϸ�ά˾Ⱥչ�ָ��Ű�ȫ�Լ�������ǰ��
����D-0502����ѧ����Taragarestrant���ǹ�˾�����з�����һ���ڷ�ѡ���ԴƼ������彵�����SERD������Ӧ֢ER+/ HER2-���ٰ���Ŀǰ��ҩ�����ڹ����������ά˾Ⱥ��ͷ��ͷ����ע���ٴ�III�����飬�з����ȴ��ڹ��ڿڷ�SERD����������ݶӡ�
����2023��SABCS�ϣ���˾����D-0502��ҩ���ƵĢ�b�ڣ�NCT03471663��������չ�εİ�ȫ�Ժ���Ч�����ݡ�
����1�������ߣ�����2023��4��7�գ�����60������HR+/HER2-�ֲ����ڻ�ת�������ٰ���Ů�����������飬�������˼���Ϊ400mgQD��D-0502��ҩ���ơ����ߵ���λ����Ϊ57�ꡣ27�������������ܹ���2�ߵ��ڷ������ƣ�27���������ܹ����ơ���36��������ESR1״̬�����������У�11��Я��ESR1ͻ�䡣
����2����Ч�ԣ���51�����������������У�8�������߲��ֻ��⣨PR����27�����������ȶ���SD��������16��SD����ʱ���24�ܡ��ٴ������ʣ�CBR��Ϊ47.1%���ۻ����ʣ�ORR��Ϊ15.7%�����������ʣ�DCR��Ϊ68.6%��
����3����ȫ�ԣ������������ز����¼���TRAE��Ϊ1-2������4/5��TRAE����������ģ������ʡ�15%��TRAE��Ż�¡�ͷ�Ρ����ġ����Ŷ����ᰱ��ת��ø���ߡ������ᰱ��ת��ø���ߡ�ƶѪ��ʳ�����ˡ���к�����ܡ����ز����¼��ķ�����Ϊ6.7%��4/60����
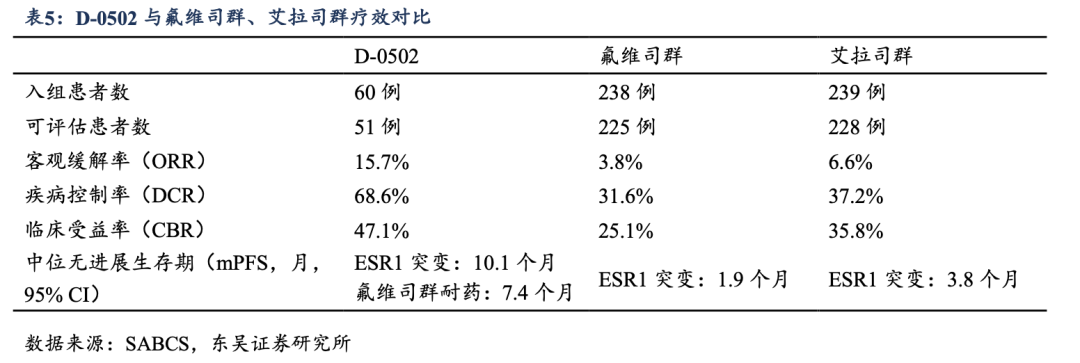
����D-0502��ҩ������HR+/HER2-��Ů�����ٰ������У������Ψһһ������SERD��ά˾Ⱥ��ȣ��ڻ������Ƶ�����£���չ�ֳ����õİ�ȫ�ԺͿ���������ǰ����Ŀǰ�������Ʒ��������ڢ����ٴ�����Σ������ڹ����ڷ�SERD������ʵ����ҵ����
����2.3. D-0120����ѡ����URAT1���Ƽ����ٴ�ǰ������֤BICDZ��
����ʹ�缰������Ѫ֢������Ⱥ���Ӵ��ݡ��й�������Ѫ֢��ʹ������ָ�ϣ�2019�������й�������Ѫ֢�����廼����Ϊ13.3%����������ԼΪ1.77�ڣ�ʹ�����巢����Ϊ1.1%����������Լ1466������Ѫ֢�ѳ�Ϊ��������Ѫѹ����Ѫ֢֬��ġ����ĸߡ�������Ҷ��-��ʪ��ѧ��������ʾ��ȫ��ʹ�综��������2020���Ѵ�5580����Ԥ�Ƶ�2050�꽫������9580���ֳ���������̬�ơ����������й�����Ⱥ������������ữ���ƣ�����50%��ʹ�综��������18��35��֮�䣬�߱�������������
������ǰ�ٴ������ֶδ������������ԣ��г�������ҩ�������Ϊ���С�������ʹ����ٴ������ֶ���Ҫ�۽�����������;�����ֱ��������������ɡ��ٽ�������й�Լ����Ƽ�����֢����������Эͬ�����˵�ǰʹ�����ƵĻ�����ϵ�������ٴ�Ӧ��Ч�����ޣ��Ҿ��������������ԣ����а�ȫ��������Ϊͻ�������������ٴ����ڹ淶���Ƽ����߸��廯����Ҳ�������Ƽ���Ԥ���Ͳ���֢�������ա�
����Ŀǰʹ��������Ҫ�������Ѵ����Dz�˾����������¡����һ��������ҩ����Ѵ������������ص�Ƥ��������Ӧ����������������Ⱥ�з��ո��ߡ��Dz�˾�����������FDA����DZ����Ѫ���������յĺڿ档����Ϊ����һ�ߴ���ҩ�������¡��DZ�ڵĸζ���δ������FDA����������2003���˳�ŷ���г������ٴ�Ӧ���趨�ڼ��ι��ܡ���Щ��ȫ������������Լ������ҩ��ij��ڡ��㷺ʹ�á����⣬�����г������»����Ŀ�ʹ����ҩ�Dz�˾������ѳ���10�꣬����ȱ����Ч�Ͱ�ȫ�Ծ�ѵ���ҩ�����»������ƴ���ʼ��ͣ��й���Ѫ�������ʽ�Լ10%��

��������ʹ�����Ƶİе��Χ��URAT1�з�������������������URAT1��Ҫ�������������С����Ƥϸ�������ֱ��������С�ܶ�����������գ��ٽ��������Һ���ų������ܾ����������й������һ���IJ������ܱ��������������ɿ��ܴ����Ĵ�л���ң��Ҷ������ܵ�Ӱ������ºͣ���ȫ�Ը������ơ�D-0120��Ϊһ���ѡ����URAT1���Ƽ���ͨ�������Ƹðе㣬�ܹ���Ч����������С��Һ�б��������ջ�ѪҺ��;�����Ӷ�ֱ�Ӵٽ�����ͨ����Һ�ų����⣬������Ҫͨ�������������ɵĻ���������ø���Ƽ��γ��˻����ϵĻ�������컯�����⣬��һ��URAT1���Ƽ����з����ļ�ͨ�����ӽṹ���Ż����ڼ̳иðе�ǿЧ������������ͬʱ��������ϵͳ�Խ��ǰ��ҩ���������ĸ��లȫ�����⣬�Ӷ����������Ƶķ��ջ���ȡ�
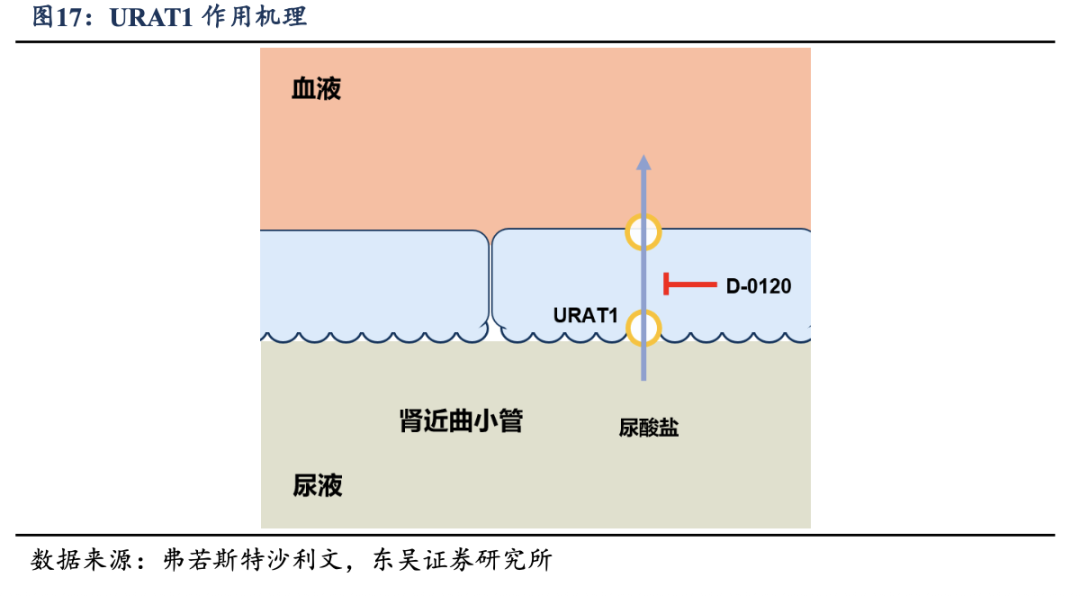
����D-0120��BICDZ���������ٴ�ǰ�о��еõ���֤�������о������ʾ���ڱ�����URAT1��ϸ��ģ���У�D-0120��URAT1�����ƻ�����������URAT1���Ƽ������ɵ£�lesinurad����150������Ч��ǿ��Ҳ������һ����ҩ��verinurad��˵��D-0120�����Ը��͵ļ���ʵ��ǿ��Ĵ�������йЧ����Ϊ��������������ƴ��ڡ�
���������ٴ�ǰ�о����ٴ�����������ʾ��D-0120��URAT1���������ý�ǿ���������ҩ������������ѪҺ������ˮƽ����������Ч����������Ӷ����ӡ��ڢ����ٴ������У�D-0120��Ʒ��������5mg��40mg�ĸ�ҩ����������������ʾ���Ϻð�ȫ�Ժ������ԣ���ʾ���Ƽ������ڽϴ��ڰ�ȫ�Է��棬�������ò�Ʒ��صIJ����¼�Ϊ1��2������ʾ�����ð�ȫ�Ժ������ԣ�����Ч�Է��棬IIa���ٴ���������ʾ���������Ľ�����Ч������ÿ�ո�ҩ4mg�����»��ߵ�Ѫ��������Ϊ80%�����⣬D-0120��Ʒ��Dz�˾����������ҩ����Эͬ���������ã�������Ч���ȵ�ҩЧ��������ǿ��ͬʱ���ٴ��о�����ʾD-0120��Dz�˾����������ҩδӰ������ҩ���ѪҩŨ�ȣ�û�����Ӷ��Ժ����ã���ʾ����������ҩ��û������á�
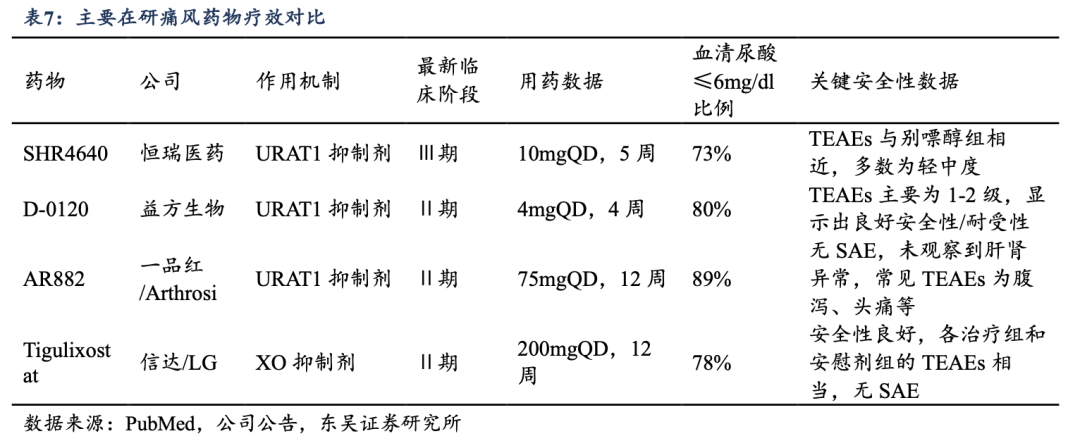
����Ŀǰ��˾D-0120����2024��11��������й���չ�����ʹ���IIb���ٴ����飬����2023��4��������������D-0120����Ѵ�������ҩ��II���ٴ���������ɻ������鼰��á�
����3. ��ҵ��������֤���۽���Сϸ���ΰ���ά����
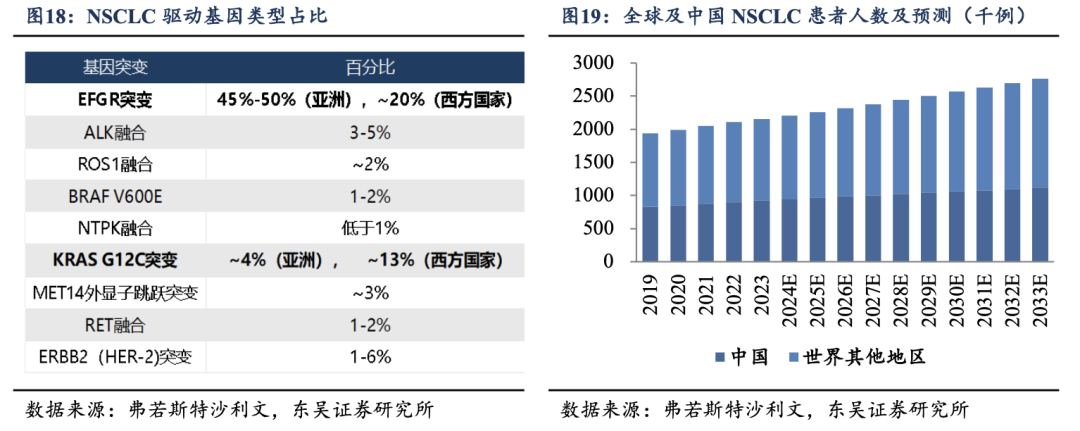
�����ΰ���ȫ�����������ʾ�����λ�Ķ����������ڷΰ�������Ⱥ�У���Сϸ���ΰ���NSCLC��Ϊ�������壬Լռȫ���ΰ�������85%����ȫ��Χ������ͬ����NSCLC���������״����������죬����EGFRͻ����KRAS G12Cͻ�乹����ߴ����Ե��������İе����ߣ��ڶ�����Ⱥ�У�EGFRͻ�䷢����ԼΪ45%�C50%�������� EGFR���������������г���������λ������ŷ����Ⱥ�У�KRAS G12Cͻ�䷢���ʿɴ�Լ13%�����Ը���������ȺԼ4%��ʹ���Ϊ����NSCLC������������ߴ����Ե�ͻ���е�֮һ��
�����ӻ��߹�ģ����ȫ���й�NSCLC�·����������������Ȳ�������ȫ��NSCLC �·�������2019���Լ194����������2024���Լ220������Ԥ��2033�꽫����Լ276������ͬ�ڣ��й�NSCLC�·�������Լ83����������Լ95������Ԥ��2033�꽫����110�����������������й�NSCLC���߹�ģ��ȫ����ռ����Ҫ���أ�Ϊ��������ҩ���ڹ���ʵ�ֳ��ڡ��ȶ���ҵ���ṩ�˼�ʵ��������
����3.1. ���������������������У���ҵ�������ƽ�������EGFR-TKI
����3.1.1. EGFR-TKI����������������
�������ݡ��й��ٴ�����Э�ᣨCSCO����Сϸ���ΰ�����ָ�ϣ�2025�棩����EGFR-TKI�ѱ�ȷ��ΪEGFR����ͻ��NSCLC�ĺ���ϵͳ�����ֶΡ�����IV��EGFR����ͻ�仼�ߣ�EGFR-TKI������һ�������Ƽ����У���һ������ TKI����ʧ�ܻ�T790Mͻ�����Ե������£�������EGFR-TKI���Ƽ����ں������ơ�
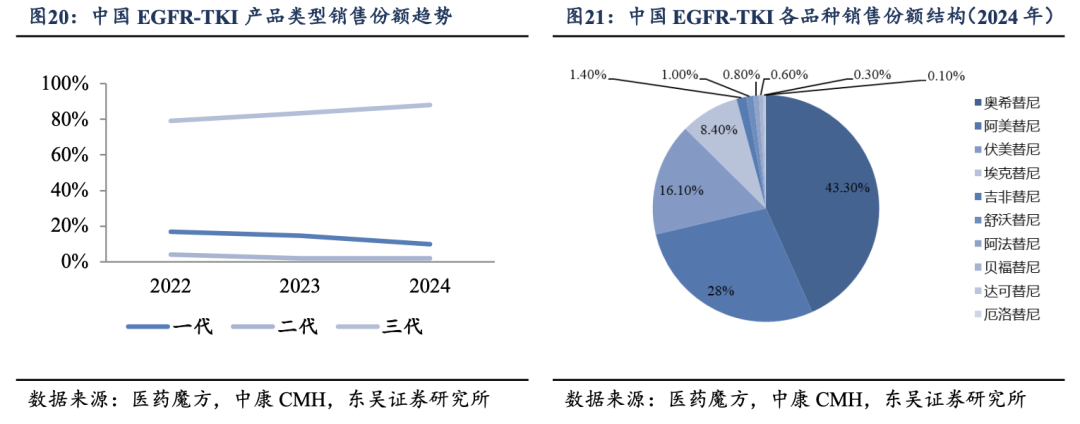
�������һ������EGFR-TKI��������EGFR-TKI��T790M��ҩͻ�����ơ���ת�ƿ��Ƽ����������Եȹؼ�ά���Ͼ߱���Ϊȫ����ۺ����ƣ������ΪEGFRͻ��NSCLCϵͳ�����еĺ�����ҩѡ����һ�ٴ�������ҩ�����ܵ�ƥ�䣬Ϊ������EGFR-TKI�ں����г�������ռ��������λ�춨�˻���������ҽҩħ�����п�CMH���ݣ�2022�C2024�������TKI����ռ����Լ79.1%������88.0%��ͬ��һ��TKI���г��ݶ���16.9%�½���9.9%������TKIռ��ѹ����Լ2%����������г��ṹ��������EGFR-TKI�����ı�Ӱ������������������������EGFR-TKI�г��ѻ�������ɡ�������桱�������������Ľṹ���л���
�����ڵ�����EGFR-TKI�ڲ������������������������ͷ���������ơ�2024���й� EGFR-TKI�г����۶���Ҫ�����ڰ�ϣ���ᡢ�������ἰ��������ȵ���������Ʒ�֣����а�ϣ�����г��ݶ�Լ43.3%�������������������ֱ�Լ28.0%��16.1%��ǰ����Ʒ�ֺϼ���ռ�ʳ���85%�����ŵ�����EGFR-TKIһ�����ʳ���������EGFR-TKI�г�������������һ��������������ͷ��Ʒ��֮�䡣
����3.1.2. �������������һ�ݶӣ��뱴������ͷ���ҵ��DZ��
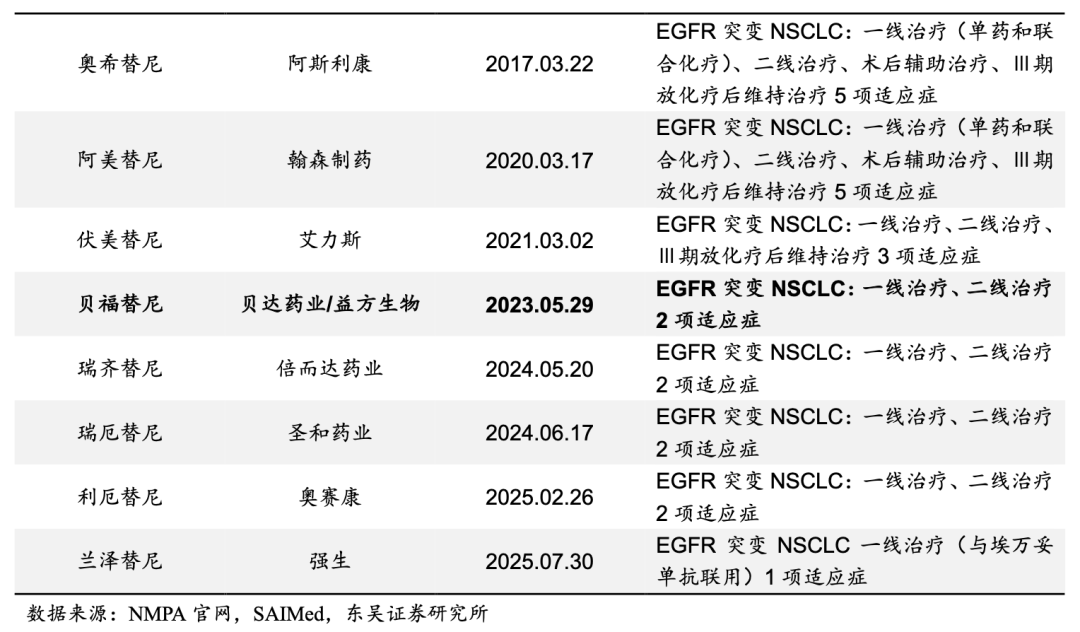
�����������ᣨBPI-D0316����Ʒ����������?���ǹ�˾�����з��ĵ�����EGFR-TKI������2023���ڹ����Ⱥ��������T790Mͻ�����ԵĶ������Ƽ�EGFR 19del/L858Rͻ���һ�����ƣ����������Ӧ֢�����������ҽ��Ŀ¼��
�������ѻ�����Ӧ֢���ֿ������ڵ�����EGFR-TKI�����ƽθ��������γ�����������ݶӽṹ����ϣ���ᡢ�������ἰ���������������һ����ҩ��������ƻ����ϣ���һ�����Ǣ��ڷŻ��ƺ�ά�ּ�/�����������Ƶȹؼ��Σ����幹�ɵ�ǰ������EGFR-TKI����Ӧ֢��������ҵ������Ⱦ��ϸߵĵ�һ�ݶӡ�
�����ڵ�һ�ݶ�֮�⣬����������Ϊ�������������еĵ�����EGFR-TKI����ǰ������ҵ�������ٷ����Σ�ͬʱ��������һ�ݶ���ҵ���Ӧ֢��չ���ԣ�����������ͬ���ƽ�EGFRͻ��NSCLC���������ƵĢ����ٴ��о������ֳ����������ƽ�ǰ�ƽ��ࡣ���֮�£��������ᡢ������ἰ��������Ȳ�Ʒ����������ʱ��϶̣���ǰ��ҵ���Դ������������Ρ�

��������Աȿ������ܵ�����EGFR-TKIȱ��ͬ��ͷ��ͷ��������о��������ڸ���ע���ؼ��ٴ��о���������������ں�����Ч�յ��ϵı����ѽ��������EGFR-TKI������ˮƽ���䡣һ�������У���mPFS����ͬ����Ʒ��������أ�����ҩ��������Ƴ����£���mPFS�밢�����ᡢ��������������к���Ʒ������ɱȣ�δ��ʾ��������Ч�̰塣�ڴ˻����ϣ�����������������ռ����ȡ������Ӧ֢��������ҵ���ƽ����ࡣ
��������������á��淽���︺�������з�������ҩҵ��������ٴ���������ҵ�����ĺ���ģʽ��2018��12�£��ڱ�����������ҩ���IND��������II���ٴ����淽�����뱴��ҩҵǩ�����Э�飬���豴��ҩҵ���й��ڵء���ۼ��й�̨������Ըò�Ʒ���п�������ҵ���Ķ���Ȩ����
��������Э�飬��˾�ɻ�úϼ�2.30��Ԫ����ҵ�����з���̱���������̱����Ʒ���й��ĺ���ע��ڵ�ҹ���ͬʱ����˾��ɻ��ڱ�������δ����Ⱦ����۶��ȡ��Ӧ����ҵ��̱�������Ȩʹ�÷ѣ�ʵ�ֶԲ�Ʒ��ҵ�������ij������롣
��������ҩҵ��Ϊ����EGFR��������������ҵ��������Ϊ����ı���ҩ��֮һ���ڷΰ�����߱����ڵ�ҽ���������г��ƹ������ͨ���뱴��ҩҵ�ĺ�����������������ڲ���������������ҵ��Ͷ���ǰ���£����г��������ӿ����к�����࣬����������һ�ߡ�������Ӧ֢���������ͷ���ҵ��DZ����ͬʱ������ҩҵ�����ƽ���������������MCLA-129��EGFR/c-MET˫�����Կ��壩���������Ʒ������ڵ�ǰհ��̽����Ŀǰ�ѽ����ٴ��������½Ρ��������ϲ����ڻ����϶Ա����ɰ�������������������������֤�Ĺ���������ʽ��Ϊ����������������Ƴ����µij���Ӧ�ñ߽��ṩDZ�ڲ��䡣
����3.2. ��������������ȷ��������ǰ��һ�ߣ���KRAS G12C�����г�
����3.2.1. ���߰���·����KRAS G12C���Ƽ�ΪNSCLC�ṩȫ�������ֶ�
����KRASͻ���ȥ���ڱ���Ϊ��Сϸ���ΰ��еġ����ɳ�ҩ���е㣬�ڽϳ�ʱ����ȱ����Ч�İ��������ֶΡ�������ȷ����Я��KRAS G12Cͻ�䣬��������������Ե�������һ�����ƾ������ݣ�����������Ʋ����ϳ��ڲ�������������NSCLC������������ϵ��һ��������Ҫ����PD-(L)1����ˮƽѡ�����ߵ�ҩ���������ϻ��Ʒ�����
��������KRAS G12C���Ƽ������ٴ������ָ���Ƽ��������Ƽ�ֵ���������ڼ�����չ��ĺ��߽Ρ�CSCO��NCCNָ���ѽ�KRAS G12C���Ƽ���������NSCLC�ĺ��������Ƽ����У�ʹ����ԭ�����ܽ���������������ƵĻ��ߣ��״λ�þ߱���ȷ���������Եİ�������ѡ��
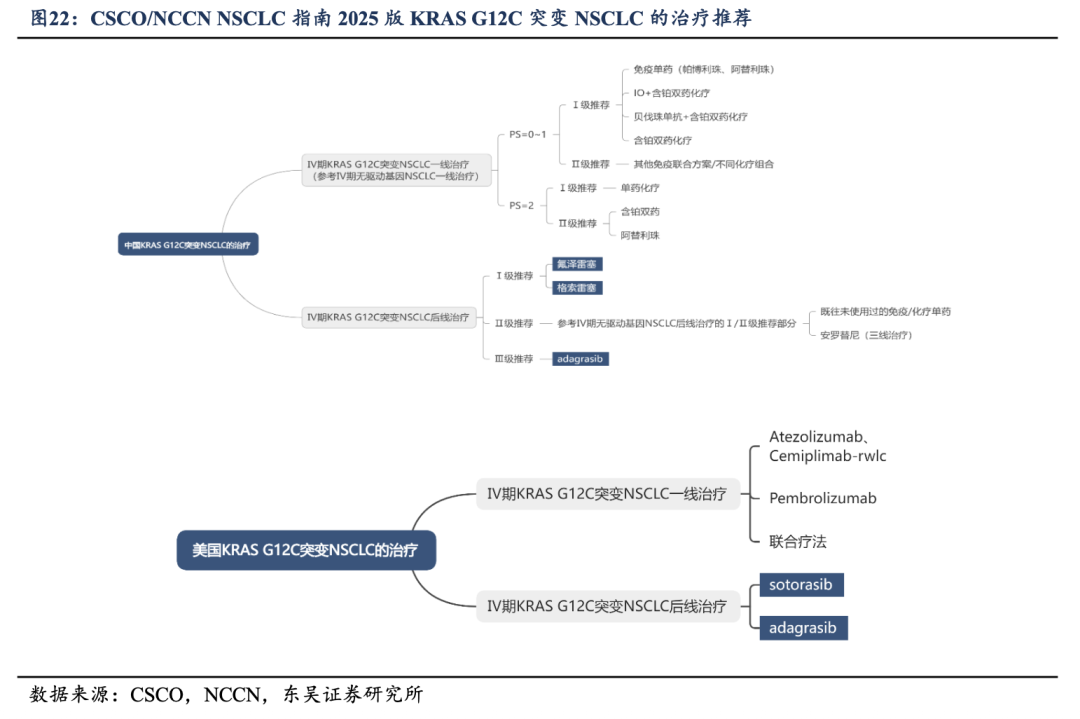
������ EGFR��ALK��ǿ��������һ�����ij�������·����ͬ���ڵ�ǰ���Ƹ���£�KRAS G12C��δȡ��һ���������Ƶ�������λ������ͨ���ں��߽�������ȷ�İ�������ѡ�Ϊ����������ϵ�ṩ�˽ṹ����������Ϊδ����������ǰ�Ƶ춨������
����3.2.2. �������������ߵ�ҩ��Ч��ȷ�������Ʒ���ǰ��CRC�ٴ�DZ��
��������������D-1553����Ʒ����������?���ǹ�˾�����з���һ��KRAS G12C���Ƽ����������ƴ���KRAS G12Cͻ��ķ�Сϸ���ΰ�����ֱ�����ȶ��ְ�֢���ǹ����������з��������ٴ�����ε�KRAS G12C���Ƽ��������������Ĵ����ǡ��й����������й�̨��ȶ�����Ҽ�������չ�˹��ʶ������ٴ����顣
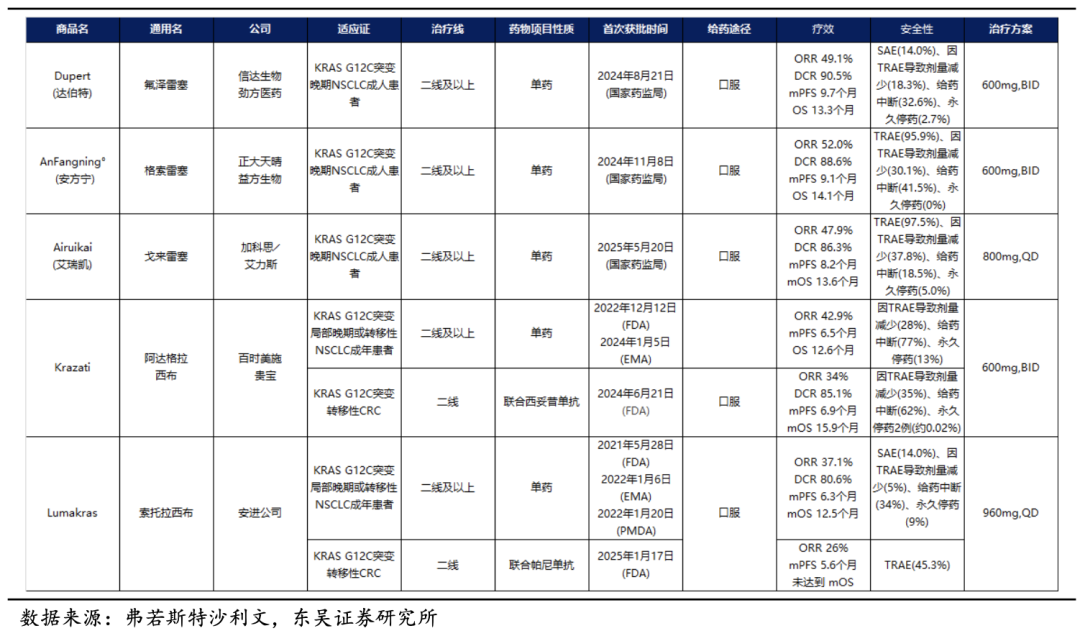
�����ڹؼ��ٴ���֤���棬��������Χ�Ƽ������ܹ�ϵͳ���Ƶ�KRAS G12Cͻ������NSCLC��Ⱥ��չע�����ٴ��о������й���չ�Ģ��ڵ����ٴ��о��У�����������ҩ������ʾ����Ϊ�Ƚ��Ŀ��������ԣ���ä̬����������飨BICR�������Ŀۻ����ʣ�ORR��Ϊ52.0%�����������ʣ�DCR����88.6%����λ��չ�����ڣ�mPFS��Ϊ9.1���£���λ�������ڣ�mOS��Ϊ14.1���£��ڰ�ȫ�Է��棬����������ز����¼���TRAE�������ʽϸߣ���δ�۲쵽��TRAE���µ�����ͣҩ���������嶾������ͬ��KRAS G12C���Ƽ�һ�£������Դ��ڿɹ������䡣�����������ݣ�����������2024��11�»����ҩ����������ڼ������ܹ�ϵͳ���Ƶ�KRAS G12Cͻ�����ڷ�Сϸ���ΰ����ߣ�2025���һ�������롶CSCO��Сϸ���ΰ�����ָ�ϡ��������Ƣ��Ƽ�������ͬ��������ҽ��Ŀ¼����ɡ�������ָ�ϡ�ҽ�����Ĺؼ��ջ���
������������KRAS G12C���Ƽ��ĺ���ȽϿ�����ǰ�������Ʒ����Ӧ֢�����������ϸ߶ȼ��У�����Ҫ��λ�ں���NSCLC��ҩ���Ƴ������ڸñ����£�����������Ϊ���ڵڶ������е�KRAS G12C���Ƽ��ں�����Чָ���ϵı����ѽ���ͬ���Ʒ������ˮƽ���䣺��ORR��mPFS�����ͬ�ڻ����ķ������������������Ȳ�Ʒ�����൱������mOSָ���ϴ���������KRAS G12C���Ƽ��е���Խϸ�ˮƽ��������ڻ����������������밢�����������ҩ���ݣ������ֳ�һ������ֵ���ơ�����������������������ɺ������Ƴ����µij���ٴ���֤��������������Ľ�һ���ͷţ��������ڼ��к��ߵ�ҩ�����ϣ�ͨ����������̽������������ǰ�ƣ��������������ռ䡣

������KRAS G12C���Ƽ���ҩ�����ѽ�����߱��������еı����£����ͻ����Чƽ̨�ڡ��ƶ���������ǰ�ƣ���Ϊ�ðе�����ٴ������ĺ��ķ���֮һ��Χ��������������ҩͨ·���������Ʋ��ԣ�����ΪKRAS G12C���Ƽ�����Ҫ����·����
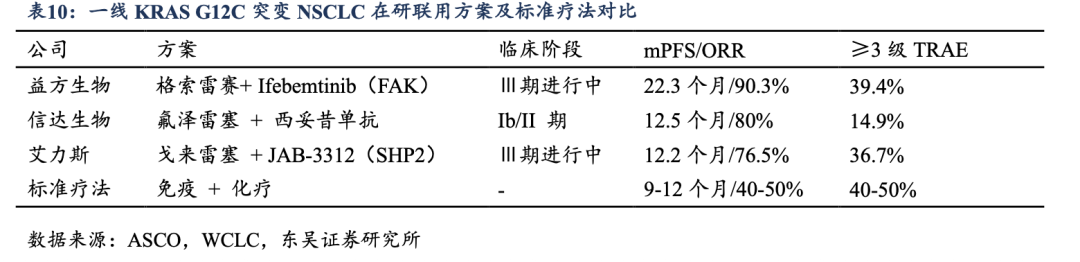
����FAK��Focal Adhesion Kinase��ͨ·��KRASͻ����������������Ϯ���������Ƽ���ҩ�γ�������ء�����FAK�źű���Ϊ�����ڸ���������������ǿ�������������ԡ��ڴ˱����£�����������Ӧ������FAK���Ƽ� ifebemtinib��IN10018�������Ϸ�����������̽��KRAS G12Cͻ��������һ�����������е�DZ�ڼ�ֵ��
������NSCLCһ�����Ƴ����У����Ϸ������ͷŻ����źš���һ��Ib/II���ٴ��о��У�����2025��3�£�II����չ���й�����33��һ��KRAS G12Cͻ��NSCLC���ߡ��������Ƶ���λ��չ�����ڣ�mPFS���ﵽ22.3���£���λ�������ʱ�䣨mDOR��Ϊ19.4���£���λ�������ڣ�mOS����δ�ﵽ����ȫ�Է���δ�۲쵽���Զ��Ե��ӣ������������뵥ҩ��������ɱȡ���س��������������2025��ASCO��ṫ����ΪKRAS G12C���Ƽ���NSCLCһ�������е�DZ��ǰ���ṩ����Ҫѭ֤���ݡ�
�����ڽ�ֱ������CRC��������������ͬ���߱���ȷ���ٴ����г�������������������KRAS G12CͻCRC��Ⱥ�л����˽�Ϊϵͳ�ĵ�ҩ�������������ݡ�1���ڵ�ҩ���Ʒ��棬2025��6�·�����Signal Transduction and Targeted Therapy�Ķ����о���ʾ������������ҩ�������ڻ�ת����KRAS G12Cͻ��CRC���ߵĿۻ����ʣ�ORR��Ϊ19.2%�����������ʣ�DCR����92.3%����λ��չ�����ڣ�mPFS��Ϊ5.5���£���λ�������ڣ�mOS��Ϊ13.1���£����ֳ����ڸ߶���ҩ�������Ծ߱�һ�����������ԣ�2������������̽�����棬����������EGFR���Ƽ����������������Ϸ���������������Чˮƽ�������о��У��������ƶ��� ORR ������45.2%��DCRΪ92.9%��mPFS�ӳ���7.5 ���£���λOS��δ�ﵽ����ʾ��ͨ��������·�ź�ͨ·�Կ˷�KRAS G12C���Ƽ���ҩ����ȷ���档
�������⣬�ڸ���ǰհ�Ե����ϲ����У�������������Ӧ������ifebemtinib����������о�����ʾ������������ڼ������ܹ����Ƶ�KRAS G12Cͻ��CRC�����У�����������ORRΪ44.4%���������ڵ�ҩ���16.7%��mPFS�ֱ�Ϊ7.7������4.0���£���һ����֤�ˡ�KRAS G12C ����+��·�ź���ϡ���CRC�е�DZ���ٴ���ֵ��
��������ҵ�����棬����������ȡ���淽���︺���з����������縺���ڵ���ҵ�����ĺ���ģʽ���������米���й�������ҩ�������ţ�����������߱������ע������ҵ����ϵ�����ڷΰ��Ⱥ���������ӵ�г���ҽ���������г��ƹ������
����2023��8�£��淽��������������ǩ�����������Э�飬���������й��ڵضԸ����������п�����ע�ᡢ��������ҵ���Ķ���Ȩ��������Э�飬�淽������Ȩ��úϼ����2.60��Ԫ����ҵ�����з���̱�����Լ����2.90��Ԫ����ҵ���ҵ����̱����ͬʱ����˾���ɻ��ڸ�������δ����Ⱦ����۶������ȡ����Ȩʹ�÷ѣ��Ӷ��ڲ��е����۷��õ�ǰ���£����������Ʒ��ҵ���������������档
����4. ӯ��Ԥ�����ֵ
����4.1. ӯ��Ԥ��
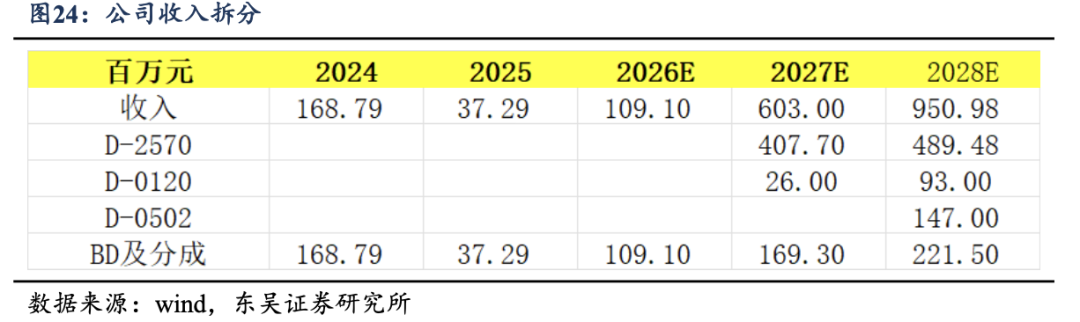
��������Ԥ�ƹ�˾2026-2028������ֱ�Ϊ1.09��6.03��9.51��Ԫ�����ļ��裺
����1��D-2570��D-0120��2027��ݽ�NDA������������У��������롣Ԥ��2027-2028��D-2570�ֱ���4.08�ڡ�4.89��Ԫ��D-0120�ֱ���0.26��0.93��Ԫ��
����2���ݲ�����D-2570�ĺ���BD����
����4.2. ��ֵ��Ͷ�ʽ���
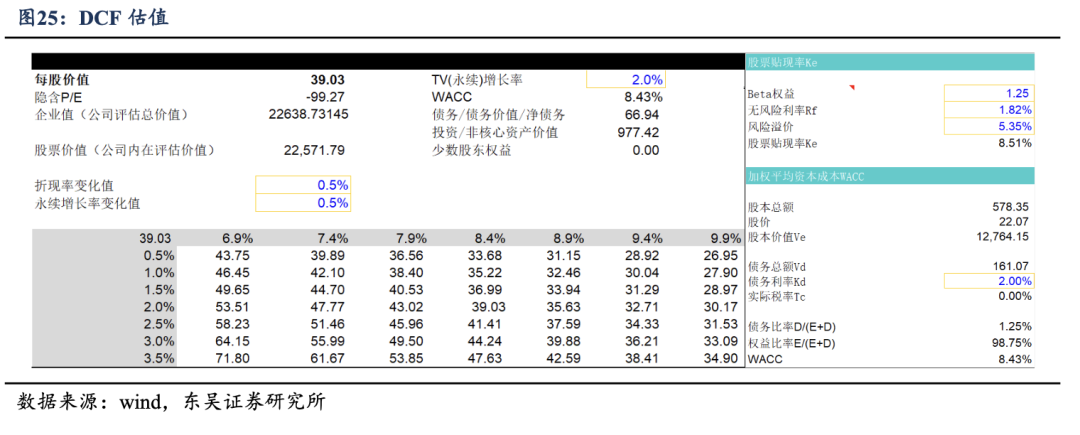
�������Dz��� DCF ���Թ�ֵ���Թ�˾���й�ֵ������Ŀ��� 39.03Ԫ���״θ��Ǹ��蹫˾�����롱������
��������
����1����˾2022��7��25�����У��ʱ���ֵѡȡ����Ϊ���е�����2026��4��17�գ�
����2���������ʲ�ȡʮ���ڹ�ծ���ʣ�
����3���г�Ԥ�������ʲο���ʮ����ָ֤���ۺ������ʡ�
����5. ������ʾ
����1���ٴ����Ȳ���Ԥ�ڣ�����˾���Ĺ���D-2570���ٴ����Ȳ���Ԥ�ڣ����Ӱ������ٴ����ȼ����н��ȡ�
����2����������ƽ�����Ԥ�ڣ�����˾���Ĺ���D-2570�ĺ���BD���Ȳ���Ԥ�ڣ����Ӱ�칫˾BD�������룬Ϊ��˾�ֽ����������գ����ӻ���Ʒ����ʱ�䡣
����3����ҵ�����۲���Ԥ�ڣ���˾Сϸ���ΰ��������ᡢ���������Ѳ�����ҵ���Σ������۲���Ԥ�����Ӱ�칫˾���롣